Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder that affects approximately 1 in 10,000 babies born worldwide each year1, 2. Deficiencies in the SMN protein result in a gradual loss of motor neurons in the anterior horn of the spinal cord and subsequent system-wide atrophy of skeletal muscles3. The gene responsible for SMA, survival motor neuron 1 (SMN1), has been identified4. A nearly identical copy of the gene, SMN2, is normally expressed in all patients with SMA. Although a small amount of full-length protein is produced that is identical to SMN1, the exon-splicing silencer bearing a C-to-T transition in exon 7 of SMN2 in all individuals skips exon 75, 6. The protein product SMNΔ7 appears to be unstable and rapidly degrades9. Increasing the copy number of SMN2 modulates the severity of SMA but does not fully compensate for the loss of SMN17, 8. Although SMN1 was identified as the mutant gene responsible for SMA 20 years ago, the molecular mechanisms by which exon 7 deletion alters cellular functions and SMA-associated mutations trigger the disease have not been elucidated.
Several misfolded disease-causing proteins harbor an intrinsic prion-like domain, which recently has been suggested to play numerous roles in normal cellular processes, such as membrane-less granule organization, alternative splicing and heterochromatin formation, underlying liquid-liquid phase separation (LLPS) via temporal homo- and hetero-cross-β polymerization (prion-like interactions)10,11,12,13,14. Similar to known prion-like low complexity (LC) proteins, SMN concentrates in subnuclear bodies called gems and is incorporated into cytosolic stress granules (SGs) through interaction with a prion-like protein, TIA115, 16. Interestingly, Lorson et al. identified a modular self-oligomerization region in exon 6 of SMN, and the disease severity was inversely proportional to the intracellular concentration of oligomerization-competent SMN proteins and the formation of gems17. Given that the membrane-less granule organization and self-oligomerization are the signature of prion-like LC proteins, we thus hypothesized that the SMN is a prion-like protein and the deficiency in prion-like functions of SMN leads to SMA. Indeed, we identified that SMN contains a prion-like LC domain at exons 6-7, which drives LLPS, and discovered an LLPS activator of gems, baicalein. We found that baicalein reinvented the C-terminus of the SMNΔ7 protein transcript encoded by SMN2 into a competent prion-like conformation, restored the prion-like function of SMN and effectively rescued SMA mice.
An Activator of SMN Associated with Phase Separation Rescues SMA
Dysfunctional targeting of SMN to gems and coiled bodies (CBs) has been suggested to be a feature of SMA in patients18. In searching for a phase separation-associated activator to rescue SMA, we found that baicalein restores the assembly of gems in SMA patient-derived human dermal fibroblasts (HDFs) by screening a chemical library (Fig. 1a). The statistical analysis of the effect of baicalein on gem body formation is shown in Fig. 1b. To determine whether the in vitro findings could be recapitulated in vivo, we treated a late-onset mouse model of SMA (type III) with daily intraperitoneal injections of baicalein (13.6 mg/kg/d) or DMSO (control). After 4 weeks of treatment, histological analysis of the lumbar spinal sections of SMA mice confirmed an increase in ChAT-positive motor neuron numbers (Fig. 1c, P=0.0435) and recovery of nuclear gems (Fig. 1c, bottom, labeled by arrowheads, P=0.0063).
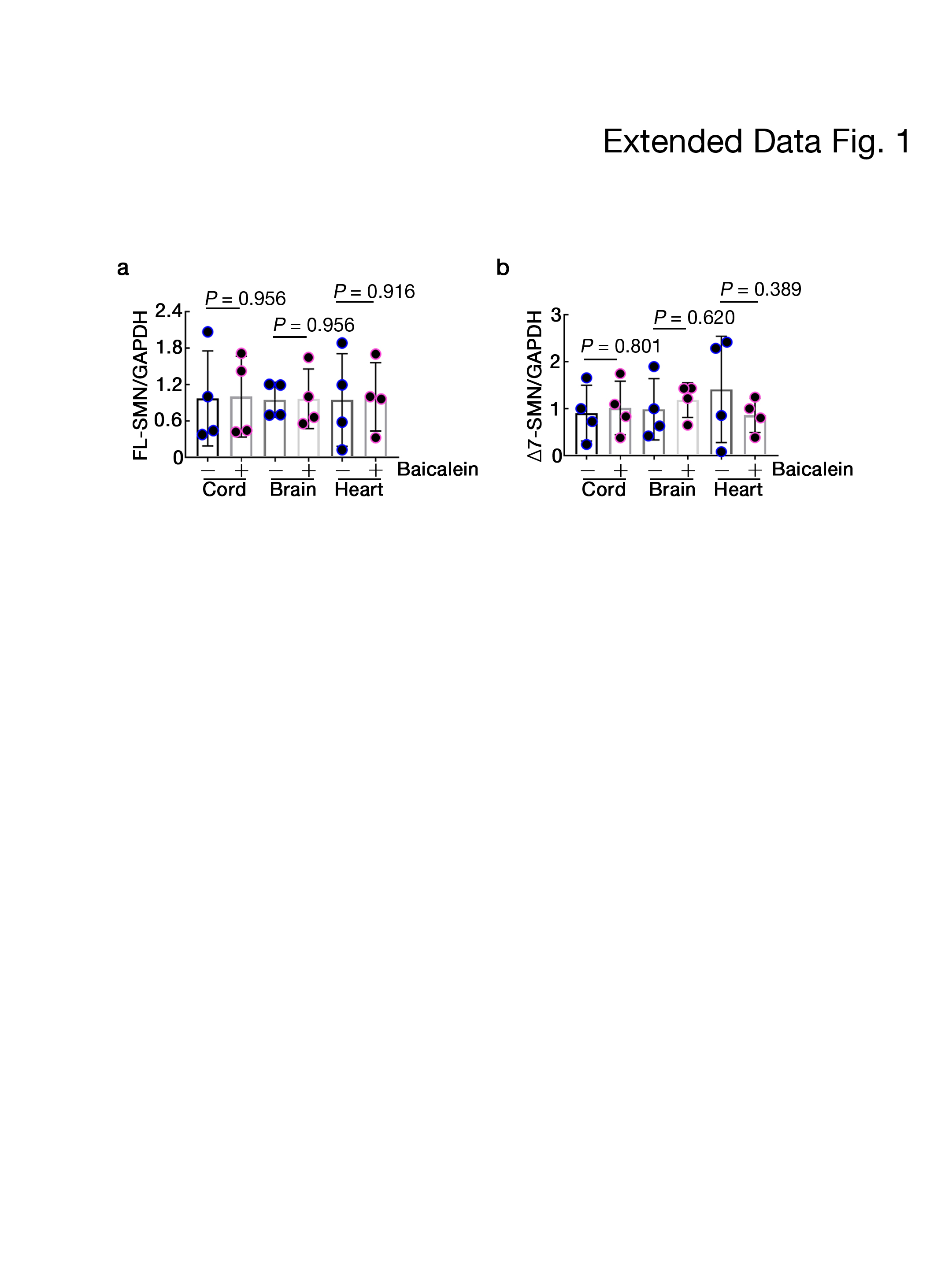
We found that the SMN protein levels were increased in various tissues of baicalein-treated SMA mice compared with those in control SMA mice (Fig. 1d, e), but the SMN mRNA levels were not (Extended Data Fig. 1), affirming that baicalein may increase the protein stability of SMND7. Beginning at 7 months of age, we next treated the type III SMA mice with baicalein for four months, which improved mouse survival (P=0.048) (Fig. 1f). A reduction in the evoked compound muscle action potential (CMAP) amplitude upon sciatic nerve stimulation was observed in control SMA mice (Fig. 1g, left, P=0.0201), but this phenomenon was prevented in baicalein -treated SMA mice (Fig. 1g, right, P=0.3326). Baicalein-treated SMA mice showed better motor performance than control mice in both the fixed-speed and accelerating rotarod tests, measuring the latency to fall (Fig. 1h and i). Fluorogold retrograde tracing of spinal motor neurons revealed a higher motor neuron density within the anterior horn (Fig. 1j, top, P=0.0114) and more large a-motor neurons (Fig. 1j, bottom, P=0.0037) in baicalein-treated mice than in control SMA mice. Significantly, baicalein treatment improved the axonal innervation of neuromuscular junctions (NMJs) in the hamstring muscles of SMA mice (Fig. 1k, P=0.04).
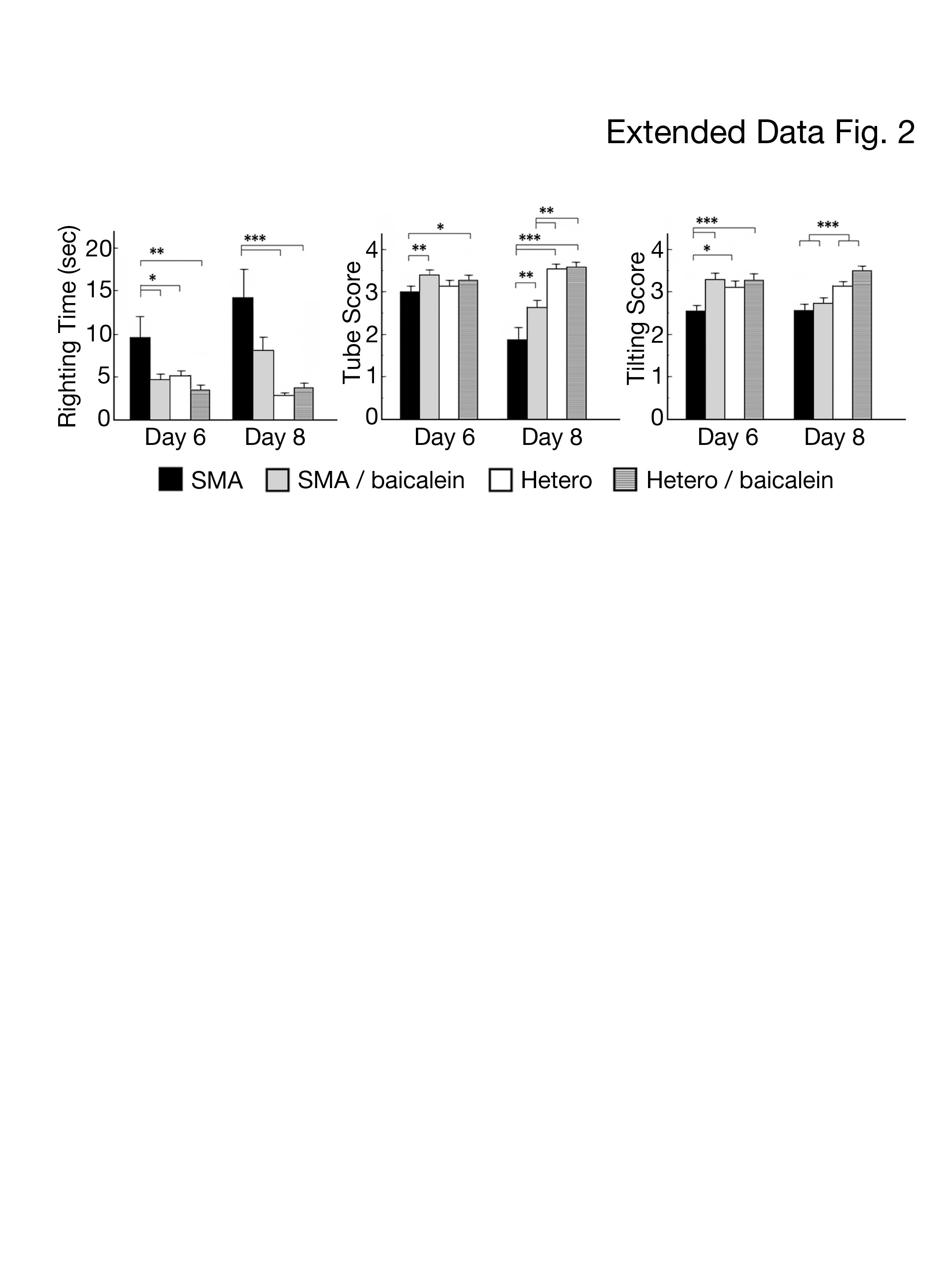
We also treated an early-onset mouse model of SMA (type I) with daily intraperitoneal injections of baicalein (13.6 mg/kg/d) or DMSO (control) beginning at birth. After baicalein treatment, the functional performances of the SMA mice, including their righting times, tube scores, and tilting scores, were improved on the 6th postnatal day (P<0.05), and the results were similar to those of their heterozygous littermates treated with or without baicalein. On the 8th postnatal day, the functional performances of the baicalein-treated SMA mice were better than those of the untreated SMA mice in the tube test (P=0.009) but not in the turnover test (P=0.065) or negative geotaxis test (P=0.58) (Extended Data Fig. 2). However, baicalein-treated type I SMA mice showed lifespans (8.8±0.4 vs. 7.9±0.2 days; P=0.13) and body weight gains that were similar to those in control SMA mice. We assumed that the limited therapeutic impact of baicalein on type I SMA mice may have been due to the shortened treatment duration.
Liquid-liquid Phase Separation of SMN
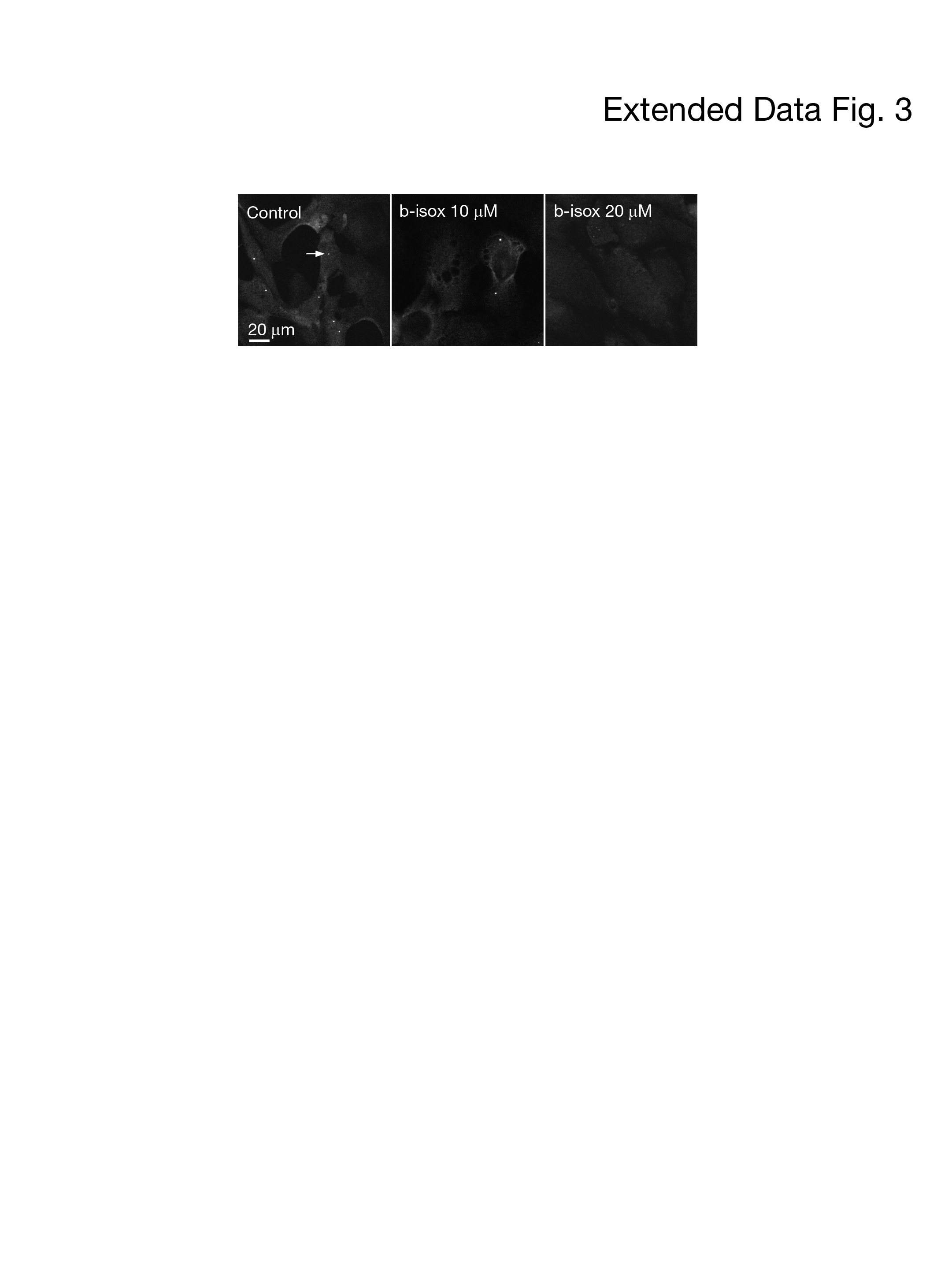
Biotinylated isoxazole (b-isox), a recently identified specific chemical probe, specifically recognizes the cross-β prion-like polymer and sequentially precipitates with proteins harboring prion-like, low-complexity or phase-separated domains, such as TDP-43, Fus and tau11. To determine whether SMN is a prion-like LC protein, we initially incubated 100 mM b-isox with lysates of mes23.5 and 293T cells at 4°C to chemically precipitate whole prion-like proteins and then analyzed the efficiency of SMN binding by Western blot to assess the prion-like and phase transition potentials of SMN (Fig. 2a). Western blot analysis revealed the precipitation of SMN by b-isox (Fig. 2a). Subcellular fractionation analysis further revealed that the major conformation of SMN recognized by the b-isox was localized in the cytosol (Fig. 2b). SET and PFN1 were used as fractionation controls (Fig. 2b). The sumoylated SMN proteins known to associate with the formation of gems, were the predominant SMN conformations detected in nuclear soluble and insoluble urea fractions19. H3K4me3 proteins were used as controls for insoluble loading (Fig. 2c). The insoluble sumoylated proteins of SMN disappeared following b-isox treatment (Fig. 2c). Consistently, we treated healthy HDFs with b-isox and found that the number of gems was significantly reduced in a dose-dependent manner, confirming that prion-like LC interactions of SMN constitute gems (Extended Data Fig. 3). We deduced that sumoylation is a cellular regulatory mechanism that triggers the separation phase of nuclear SMN though cross-β polymerization.
To determine prion-like domain of SMN, we examined the LLPS capabilities of full-length SMN (SMN-FL) and a panel of exon-deleted SMN constructs. The map of the SMN variants and results are shown in Fig. 2d. Only fragments containing the region encoding exons 6-7 formed visible granules (Fig. 2e, arrowhead). The dynamics of mCherry-SMNexon6-7 granules in living cells were further analyzed by time-lapse microscopy, and the trajectory of mCherry-SMNexon6-7 granules is shown in Fig. 2f. Most of the mCherry-SMNexons6-7 granules traveled within a limited location (Fig. 2f), and two independent granules of mCherry-SMNexon6-7 occasionally underwent fission and fusion (Fig. 2g, arrowheads). The average granular speed of mCherry-SMNexon6-7 was ~0.8 µm/s. As the prion-like domain of SMN is localized at exons 6-7, we inferred that the deletion of exon 7 leads to insufficient prion-like activity of SMN protein.
Prion-like Conformational Editing of SMNΔ7 Proteins by Baicalein
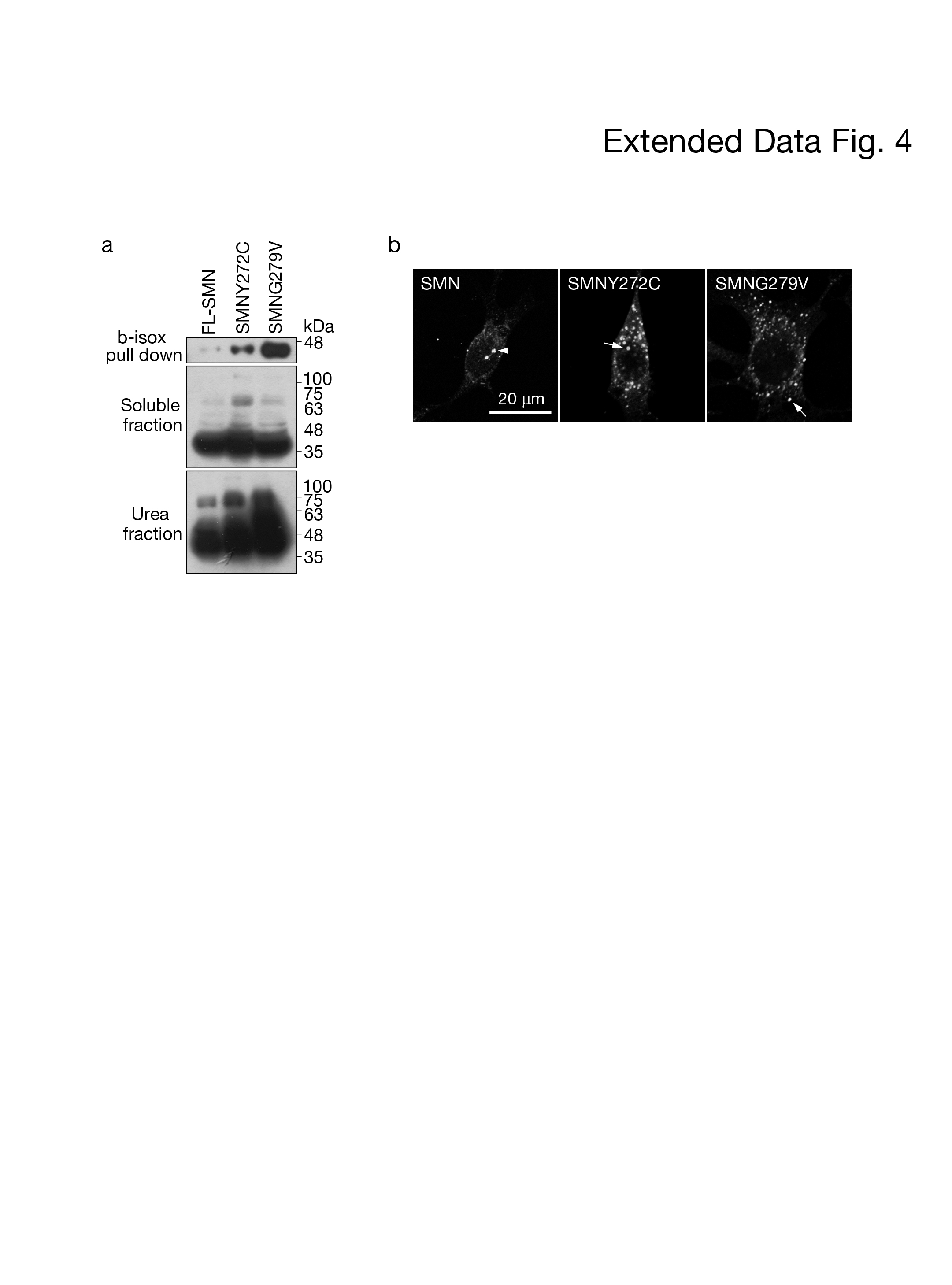
As the SMA patient-derived HDFs and SMA type III mice shown in Fig. 1 express only the SMN2 proteins, we speculated that baicalein restores gems in SMA patient-derived HDFs and SMA type III mice by editing the conformation of the C-terminus of SMNΔ7. Using the b-isox precipitation assay to detect the in vivo cross-β conformation of SMN, we found that the cross-β conformers of SMN in HDFs and SMA patient-derived HDFs were different (Fig. 3a). Importantly, the cross-β conformers of SMNΔ7 in SMA patient-derived HDFs were recapitulated as cross-β conformers of SMN-FL upon baicalein treatment (Fig. 3a). This result supported that baicalein can edit the conformation of SMNΔ7 into a prion-like conformer of SMN1 and explain the pharmacological mechanism of baicalein in restoration of the formation of gems in Figure 1a and 1c. In order to assess whether the prion-like activity of SMNΔ7 per se increases upon baicalein, we performed coimmunoprecipitation to analyze the prion-like interactions of SMNΔ7 and PFN1. PFN1 is a aggregation-prone protein co-precipitates with b-isox, and known to effectively interact with SMN but not SMNΔ7 (Fig. 2b) 20, 21, 22. The results showed that in the presence of baicalein, the interactions between PFN1 and SMNΔ7 were significantly increased (Fig. 3b). As the prion-like conformation is critical for the protein stability of TDP-43, we next examined whether baicalein attenuated the degradation of the SMNΔ7 protein, and it indeed significantly reduced SMNΔ7 degradation (Fig. 3c, arrow). In a previous study, neurons transfected with SMNΔ7 extended significantly shorter neurites than those transfected with SMN-FL23, 24. Baicalein increased the length of axons from NSC34 cells expressing SMNΔ7 by approximately 2-fold (Fig. 3d); the statistical analysis is shown in Fig. 3e. These results together with those in Fig. 1 suggest that baicalein endows SMNΔ7 with prion-like bioactivity. We notice that two patient-derived missense SMN1 mutants, Y272C and G279V, mutated in exon 6 increased the binding affinity of the b-isox and the insolubility. Change in these two biochemical properties reflected that their cellular conformation was altered (Extended Data Fig. 4). These results suggested that amino acids 272 and 279 and exon 7 are critical for the adoption of a competent prion-like conformation of SMN, and the impaired prion-like activity of SMN1 is the root cause of SMA.
To validate the role of prion-like activity in SMA, we increased the amount of functional prion-like domains by overexpressing the TDP-43 prion-like domain (TDP-43-PLD) in NSC34 motor neuron cells expressing the SMNΔ7 protein to confirm that the level of the functional prion-like conformer affects axon degeneration. TDP-43-PLD is expected to adopt a common, structurally similar cross b-sheet to compensate for the prion-like function of SMN. Our experiment showed an increase in axon length in cells expressing both SMNΔ7 and GFP-TDP-43 PLD compared to the GFP- and GFP-NPLD-expressing controls (Fig. 3f, arrowheads). The statistical analysis is shown in Fig. 3g. Conversely, SMN overexpression in motor neurons has also been shown to slow the onset of amyotrophic lateral sclerosis (ALS) and pathological symptoms in a model of mutant TDP-4325, 26, 27. Thus, the level of the prion-like conformer is critical for motor neuron survival, and other functionally unrelated prion-like proteins can compensate for the function of the defective protein.
Based on the unique role of prion-like conformations in motor neurons, we propose a therapeutic model of SMA, “the prion-like conformation-based therapies”, in which SMA disease-causing mutants and SMNΔ7 are converted into prion-like folded proteins (Fig. 3h). Baicalein endowed SMN mutants and SMNΔ7 with prion-like activity, subsequently triggering LLPS of SMNΔ7 to form gems and increasing prion-like interactions, i.e. SMNΔ7-PFN1, reducing protein degradation of SMN, and improving motor neuron survival and motor functions in SMA patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}