Materials
Chloroauric acid, tetrahydrate (HAuCl4·4H2O, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). DTX and PSMA antibody were purchased from Sigma-Aldrich (St. Louis, USA). DUPA (OtBu)-OH was purchased from Med Chem Express (New Jersey, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Japan). Deionized water with resistivity of 18.2 MΩ·cm was used in all of the experiments. All agents were used without further purification.
Synthesis of Au@DTX-DUPA NPs
Au NPs were prepared by the typical reduction method[17] (Supporting information). For Pegylated Au NPs, excess of SH-PEG2K-NH2 and SH-mPEG2K ligands in ratio of 1:4 molar was used to react with 1 equiv of naked Au NPs for 12 h. To visualize the PEG corona, the PEGylated Au NPs samples were stained with 2% phosphotungstic acid prior to TEM observation. Pegylated Au-DUPA NPs was synthesized as previously reported with minor modifications.[18, 19] Briefly, DUPA(OtBu)-OH (0.2 mmol) was dissolved in dichloromethane (10 mL), followed by the addition of Hydroxybenzotriazole (1 equiv), EDC-HCl (2 equiv), and triethylamine (1 equiv), in ice bath for 1h. Subsequently, Pegylated Au NPs modified with amino terminal PEG (0.01g) dispersed in dichloromethane (5ml) were added to mixed system, the reaction mixture was stirred at room temperature for 24 h in a N2 atmosphere. Then, the protected group of DUPA(OtBu)-OH were removed by the addition of trifluoroacetic acid (TFA, 50%). Next, the organic solvent was removed by reduced pressure distillation to yield Au-DUPA NPs. Excess ligands and other reaction products were removed by extensive purification (MWCO = 30 kDa) for 3 times. The purified Au-DUPA NPs were collected and followed by freeze-drying.
DTX Loading and Release
DTX was encapsulated into PEGylated Au-DUPA NPs through the non-covalent interaction to construct the Au@DTX-DUPA NPs. First, DTX was added to Au-DUPA NPs in dichlormethane solution with gradient weight ratio (DTX/Au-DUPA NPs = 1:50, 1:25, 3:50, 1:12.5, 1:10, 1:8.3, 1:7.1, 1:6.25). Then, the mixture was slowly stirred at room temperature for four days. After dichlormethane was completely evaporated, the products were re-dispersed into 10 mL DI water for further UV-Vis analysis. The typical band of free DTX in water was set as reference. The disappearance of the typical band of DTX indicated that there was no free DTX in the supernatant and almost all DTX molecules were encapsulated into the PEG coronal of Au-DUPA NPs. In order to evaluate in vitro DTX release from Au@DTX-DUPA NPs, HBSS was chosen as simulated body fluid. In brief, 2.0 mg of Au@DTX-DUPA NPs was loaded into a dialysis tubing (retention molecular weight 12 kD) and then the dialysis tubing was maintained in 40 mL HBSS in a 37℃ water bath shaker. At designed time interval, 0.5 mL HBSS was obtained for DTX detection and another 0.5 mL fresh HBSS was supplemented. For DTX detection, the obtained releasing mixture were extracted with dichloromethane twice. Then 0.5 mL methanol was added to dissolve DTX for further high-performance liquid chromatography assay. The mobile phase was acetylene and the chromatography column was Agilent ZORBAX Eclipse XDB-C18 (5 μm).
Cell Culture and In Vitro Cytotoxicity
Human PCa cell lines (PC-3, 22RV1) and mouse fibroblast cell line (L929) were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences and maintained in RPMI-1640 medium supplemented with 10% Fetal Bovine Serum, 50 μg/mL penicillin, 50 μg/mL streptomycin, and 2 mM L- glutamine. The biocompatibility of Au-DUPA NPs was evaluated with L929 cell line. L929 cells were seeded in 96-well plate and grown for 24 h. Then, cells were incubated with cell culture medium containing different concentrations of Au-DUPA NPs for 24 and 48 h, respectively. Next, the cells survival was calculated by CCK-8 assay. To compare the cytotoxicity of Au@DTX-DUPA NPs and free DTX and identified EC50 values of two agents for 22RV1 cell. 22RV1 cells were seeded in 96-well plate and grown for 24 h. Then, cells were incubated with cell culture medium containing different concentrations of Au@DTX-DUPA NPs and free DTX for 24 h. Next, the cells viability was determined by CCK-8 assay. The dose-effect curves of Au@DTX-DUPA NPs and free DTX were built and the EC50 of two agents for 22RV1 cells were identified, Absorbance was measured at 450 nm using 96-well plate reader (Varioskan Flash, Thermo).
Cellular Uptake of Au-DUPA NPs
First, the expression of PSMA on 22RV1 and PC3 cell lines were determined by immunohistochemistry. Then, two kinds of cells were incubated with 30 μg/mL FITC-labeled Au-DUPA NPs for 24 h in 6-well plates. Then, the cells were washed with HBSS (pH = 7.4) twice and stained with Hoechst 33342 (2 μg/mL) for 10 min at 37℃. Afterward, the cells were washed with HBSS again, and observed by confocal laser scanning microscope. All images were collected on a Nikon laser scanning confocal microscope system with a 20X objective. The FITC-labeled NPs were detected by an excitation wavelength of 488 nm and an emission wavelength of 535 nm. Hoechst 33342 was activated by an excitation wavelength of 343 nm and an emission wavelength of 483 nm to label nucleus. The intracellular content of Au was quantified by ICP-MS (NexION 300 D, PerkinElmer Corporation, USA).
Detection and Quantification of Cell Cycle and Intracellular NPs Concentration
The effects of various treatments on the cell cycle distribution were analyzed by FACS (BD FACSARIA, Bio-Rad). The 22RV1 cells were incubated with Au-DUPA NPs, free DTX, or Au@DTX-DUPA NPs. After indicated incubated periods (8, 16, 24 h), the medium was removed, and 22RV1 cells were harvested by trypsinization, centrifugation, and fixed in 70% ice-cold ethanol at 4 ℃ overnight. Afterward, the cells were washed, re-suspended and treated with 10 μg/mL RNase (Sigma–Aldrich Co., US) for 30 min at 37 ℃, and then stained with HBSS containing 50 μg/mL propidium iodide (PI) for 30 min at 4℃. Finally, the stained cells were analyzed by FACS. As for NPs concentration, cells were incubated with indicated concentration of FITC-labeled Pegylated Au NPs, Au-DUPA NPs, Au@DTX-DUPA NPs, respectively. After indicated incubated periods (8, 16, 24 h), the medium was removed and cells were harvested. Cell suspension was centrifuged, and the supernatant was removed. After that, the cell samples were resuspended by HBSS and were submitted to the procedure for measuring average FITC fluorescence intensities using FACS.
Evaluation of Radiosensitive Efficacy In Vitro
The enhancement of radiotherapy efficacy in vitro was evaluated by apoptosis related protein expression and colony formation assay. 22RV1 cells were incubated with indicated concentration (30 μg/mL, with respect to Au) of Pegylated Au NPs, Au-DUPA NPs, Au@DTX-DUPA NPs, respectively for 24 h. Then, 4 Gy IR was given. Cells without treatment were set as control. Cells were collected and lysed with RIPA buffer. The supernatant was collected after centrifugation at 16,000 g at 4 ℃ for 15 min. Protein concentration was measured with BCA method. Then protein lysis buffer was treated with 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes for western blot analysis, then blocked with 5% non-fat milk, and probed with primary antibodies at 4 ℃ overnight. The primary antibodies are as following: Cleaved PARP (1:1000, CST), β-Actin (1:5000, Abcam). The membrane was washed with TBST 3 times, and incubated with HRP linked secondary antibodies for 1 h at room temperature, followed by washing with TBST. Then, the membrane was incubated with ECL (Bio-rad) for 5 min. Protein band was visualized by using a Bio-Rad Chemi-Doc MP imaging system. For colony formation assay, 22RV1 cells were seeded in 6-well plates at a concentration of 1*104/ well and incubated with indicated concentration of Pegylated Au NPs, Au-DUPA NPs, or Au@DTX-DUPA NPs for 24 h. Then, the medium was removed and cells were washed with HBSS to remove the non-internalized NPs. 4 Gy IR was given to each group. IR only group was set as reference Next, the cells were trypsinized, counted, and seeded into 6-well plates. After incubating for 12 days, the colony formation was washed with HBSS and fixed with 4% paraformaldehyde and 0.4% crystal violet solution in HBSS was added to stain the colonies. Then the number of them were counted to calculate the surviving fraction.
Detection and Quantification of ROS
22RV1 cells were seeded into 6-well plates and incubated with indicated concentration (30 μg/mL, with respect to Au) of Pegylated Au NPs, Au-DUPA NPs, Au@DTX-DUPA NPs for 24 h. Cells received IR only were used as reference. After incubation, the medium was removed and cells were washed with HBSS. DCF-DA agent (50 μM) in serum-free medium was added to each well and incubated for another 40 min. Then, the cells were washed with HBSS twice and received under 4Gy IR. Next, the cells were trypsinized, resuspended in HBSS. The average DCF-fluorescence intensity in cells were measured by FACS. As for imaging DCF-fluorescence, cells cultured in 96-well plates were washed with HBSS, added DCF-DA agent, radiated, and imaged with confocal laser scanning microscope by sequence.
Detection and Quantification of DSBs
22RV1 cells were incubated with indicated concentration (30 μg/mL, with respect to Au) of Pegylated Au NPs, Au-DUPA NPs, Au@DTX-DUPA NPs for 24 h, before being subjected to 4Gy IR. Cells with IR only were used as reference. After IR, the cells were trypsinized and immersed in 4% paraformaldehyde fixation for 15 min and then washed with HBSS three times. After that, treating the cells with Triton X-100 for 30 min to damage their membranes. Next, the cells were soak in a blocking buffer (1% bovine serum albumin in tris-buffered saline solution) for 1 h and incubated with antihistone γH2AX mouse monoclonal antibodies (diluted 1:500 with HBSS) in the dark overnight at 4 °C, followed by washing with HBSS three times. Then, they were further cultured with sheep anti-mouse secondary antibody (diluted 1:2000 with HBSS) for 2 h in dark and then washed with HBSS three times. Afterwards, 10 μL Hoechst 33342 was used to stain the cells for 15 min. Finally, the stained cells were analyzed with FACS. As for imaging the fluorescence, cells in 96-well plants were washed twice with cold HBSS after IR, fixed with 70% ethanol for 2 h, then treated with Triton X-100, soaked in a blocking buffer, incubated with antihistone γH2AX mouse monoclonal antibodies, cultured with sheep anti-mouse secondary antibody, and observed with confocal laser scanning microscope by sequence.
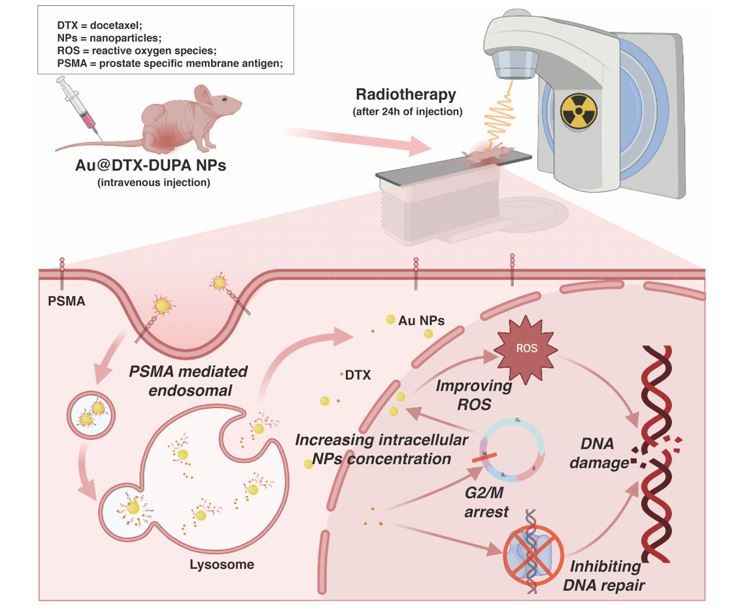
Biodistribution of NPs and In Vivo Anticancer Efficiency
Xenograft tumors were inoculated in the right flank of male athymic nude mice (6 weeks old, 18-23g) by subcutaneous injection of 1 × 106 22RV1 cells in 100 μL of 1:1 (v/v) mixture of serum-free RPMI-1640 and matrigel solution. When tumor size reached about 100 mm3, tumor-bearing mice were divided randomly into 5 groups: control, RT only, Au NPs + RT, Au-DUPA NPs + RT, and Au@DTX-DUPA NPs + RT group. NPs were given to mice with the dose of 5 mg/kg (with respect to Au). 24h after injection of NPs, the mice received 6Gy electron beam under Varian True Beam system (SN1698). Machine operating at 6 MeV, 20 cm source subject distance, and 50 cGy/min dose rate. Only the tumor region was irradiated, as the remaining parts of the body were lead-shielded. All treatments were carried out only once. Tumor sizes and body weight were measured every day. Tumor volumes were calculated using the formula V = 0.5 *a *b2, where “a” and “b” are the length and width of tumor, respectively. The data were collected until 14th day. Then, the mice received isoflurane anesthesia and were sacrificed by cervical dislocation. To evaluate the biodistribution of NPs, vital viscus (liver, lungs, heart, kidneys, spleen) and xenograft tumors of mice were collected at 12 h post injection of Au@DTX-DUPA NPs. Before analysis, all the tissue samples were stored at −20 °C. Then, the samples were thawed, weighed, grinded, and then digested in 3 mL of aqua regia at 65 °C overnight. Finally, the digested samples were diluted up to 20 mL with DI water and analyzed by ICP-MS.
Statistics
All experiments were performed in triplicates unless stated otherwise. All analyses were performed using R version 3.6.1 software (The R Foundation for Statistical Computing, Vienna, Austria; www.r- project.org). All numerical results are expressed as mean SD. Descriptive statistics and significant differences between groups were analyzed using two-tailed Student's t-tests, and the difference was considered significant if *p < 0.05 and **p < 0.01.
{kind=link}