Materials. Human IGF-I was purchased from PeProTec (UK). Primary antibodies were monoclonal anti-IGF-I receptor (1:1000; Santa Cruz Biotechnology, USA), monoclonal anti-APP (Nt 22C11; Millipore; 1:200), for PLA studies, polyclonal anti-APP (Sigma; 1:200), for immunoprecipitation, and monoclonal anti-pTyr (1:1000, Transduction Labs, USA). Secondary antibodies were goat anti-rabbit (1:20000) or mouse IRDye-coupled (1:20000), both from LI-COR (USA).
Animals. Male adult (3–5 months old) and new-born wild type C57BL6/J mice, and adult liver IGF-I deficient mutant mice (LID mice; bred in-house, congenic with C57/BL6/J) were used. LID mice present low levels of serum IGF-I due to the disruption of the liver IGF-I gene with the albumin-Cre/Lox system [24]. Serum IGF-I deficient mice have normal body and brain weights and they do not show any major developmental defects [24, 27]. Animal procedures followed European (86/609/EEC & 2003/65/EC, European Council Directives) and approval of the local Bioethics Committee.
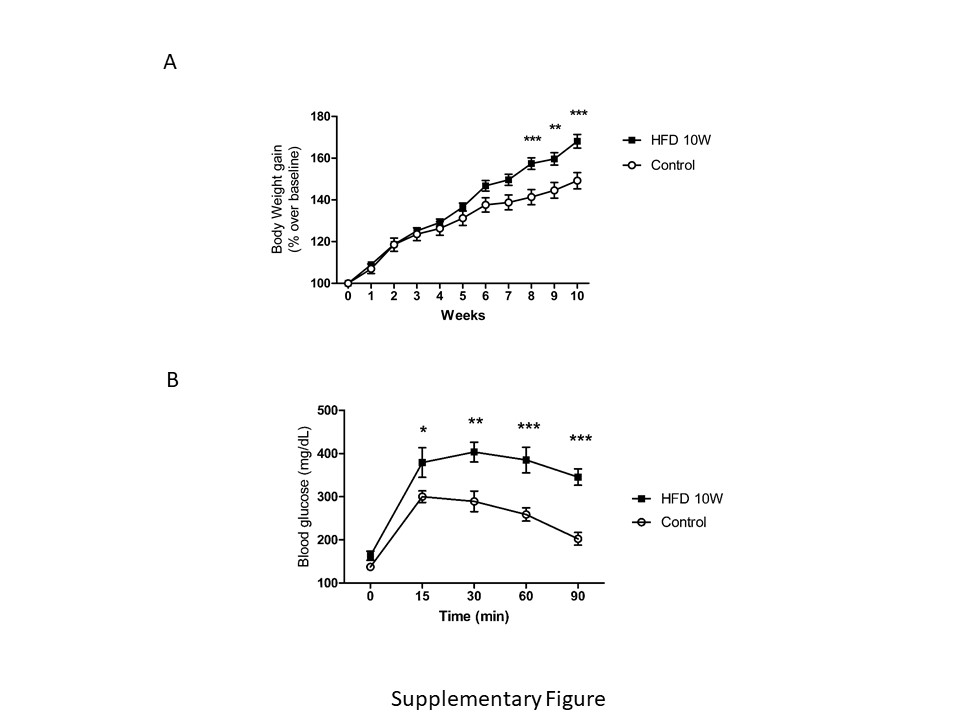
High fat diet. Wild type C57BL6/J mice were fed for 10 weeks with either a control diet (ref E15000-04), or a high fat diet (HFD) with 45% KJ fat + 1.25% cholesterol (ref E15744-34), both purchased from ssniff Spezialdiäten GmbH (Germany). After 10 weeks, animals were overweighed (Suppl Fig A), developed glucose intolerance (Supp Fig B), together with hyperinsulinemia and insulin resistance (not shown).
Cell cultures. Astroglial cultures with > 95% GFAP-positive cells were prepared as described [28]. Postnatal (day 1–2) brains were dissected, forebrain removed, and mechanically dissociated. The resulting mixed cell suspension was centrifuged and plated in DMEM/F-12 (Life Technologies) with 10% fetal bovine serum (Life Technologies) and 100 mg/ml of antibiotic-antimycotic solution (Sigma-Aldrich, Spain). When confluent, cells were shaken (210 rpm/37ºC/3 hours) to detach microglial cells. For microglial cultures, supernatants were centrifuged (1000 rpm/5 min), re-suspended in DMEM/F12 (Life Technologies) + FBS (Gibco, USA), HS and penicillin/streptomycin solution. Cells were seeded at 12.5–104 cells/cm2 in a multi-well coated with poly-L-lysine [29], and cultured for 2 days. Cells were then changed to DMEM/F12 for 3 hours until Aβ uptake was carried out (see below). Astrocytes were then collected from the same flasks that microglia was obtained, as follows. After removing the microglia-containing supernatant, the medium was replaced and flasks shaken for 15 h/280 rpm. Cells were then trypsinized and seeded at 3.75 × 104 cells /cm2 in the same culture medium, replaced every 4 days. When 80% confluency was reached, astrocytes were cultured for 3 hours with DMEM/F12 before the different assays were initiated (see below). Endothelial cell cultures were performed as described [29]. Briefly, dissection was performed on ice and cortices were cut into small pieces (1 mm3), digested in a mixture of collagenase/dispase (270 U collagenase/ml, 10% dispase) and DNAse (10 U/ml) in DMEM for 1.5 h at 37 °C. The cell pellet was separated by centrifugation in 20% bovine serum albumin/DMEM (1000 g, 5 min). Capillary fragments were retained on a 10 µm nylon filter, removed from the filter with endothelial cell basal medium (Life Technologies), supplemented with 20% bovine plasma-derived serum and antibiotics (penicillin, 100 U/ml; streptomycin, 100 µg/ml), and seeded on 60-mm Petri dishes multi-well plate coated with collagen type IV (5 µg/cm2) and fibronectin (1 µg/cm2). 3 µg/mL puromycin was added for 3 days, removed from the culture medium and replaced by fibroblast growth factor (2 ng/ml) and hydrocortisone (1 µg /ml). For hepatocytes cultures, adult (2 months old) control animals were anesthesized (pentobarbital 50 mg/kg), and the hepatic portal vein exposed to inject a solution containing NaCl (118 Mm), KCl (4.7 Mm), KH2PO4 (1.2 Mm), NaHCO3 (25 Mm), glucose (5.5 Mm), and EGTA (0.5 Mm) at 37C. The inferior cava vein was cut to open the circuit. Thereafter the same solution without EGTA and containing CaCl2 (2 Mm), MgSO4 (1.2 Mm), and colagenase (90 U/ml) was perfused. The liver was dissected and placed in DMEM/F12 -10% FBS with penicillin/ streptomycin, filtered in a 70 um Nylon mesh, centrifuged (60 g, 5 min) and re-suspended in DMEM/F12-10% FBS with 45% Percoll® (Sigma Aldrich). Cells were then re-suspended and washed 3X in DMEM/F12-10% FBS using 200 g, 10 min spins, before plating them at 8.25 x \({10}^{4}\) cells/collagen-coated multi-well. Cultures were kept 2 days before use.
Glucose tolerance test (GTT). Mice were fasted for 6 hours and left isolated in individual cages (with water but no food access) for at least 30 min before starting the test to avoid any stress-related effect on glycemia [30]. For the glucose tolerance test (GTT), an overload of glucose (2 g/kg) was injected intraperitoneally. The aqueous solution was left overnight at room temperature so the β-form of glucose was enriched. Blood samples were extracted from the tip of the tail at time 0, 15, 30, 60, and 90 to measure glucose levels with a glucometer (Menarini Diagnosis, Italy).
Environmental enrichment. Mice were submitted to environmental enrichment as explained in detail elsewhere [31]. Briefly, animals were placed for 2 hours in a large cage, 10 animals/cage and with different objects (cardboard tunnels, shelters of different materials, a plastic net, toys, chewable and nesting material). Thereafter, they were sacrificed and their brain collected for immunoprecipitation and western blot analysis.
Aβ uptake. In vitro: Cells were treated during 15 hours with 500 nM soluble Aβ40-HiLyte Fluor™ 488 (AnaSpec) [32], and IGF-I (1 nM in glial cultures, 10 nM in hepatocytes). Thereafter, cultures were washed with PBS pH 6.0 to eliminate membrane bound Aβ followed by PBS pH 7.4. Cell nuclei were stained with Hoechst 33342 (Thermo Fisher Scientific; 1:500) in PBS pH 7.4/5 min, fluorescent images were taken in an DMI 6000 (Leica) microscope using Exc: 350 nm/ Em: 461 nm for Hoeschst dye and Exc: 503 nm/ Em: 528 nm for fluorescently labeled Aβ. Thereafter, cells were lysed in Tris-HCl (10 mM) pH 8.0, guanidine (50 mM), and spinned at 14.000 rpm for 10 min at 4ºC. Fluorescence was quantified in a FLUOStar OPTIMA (BMG Labtech) at Exc: 485 nm/ Em: 520 nm. In transcytosis assays using brain endothelial cells, Aβ40-HiLyte Fluor™ 488 soluble (500 nM) was added in the bottom compartment (Fig. 4C) with or without 1 nM IGF-I, and after 15 hours the culture medium from the upper chamber was collected and fluorescence measured in the fluorimeter, as above.
In vivo
Aβ40-HiLyte Fluor™ 488 (400 µg/kg) was injected into the tail vein using a 0.38 mm cannula (Intramedic, Spain), and after 90 min mice were sacrificed, blood taken from the heart and liver dissected. Liver tissue was homogenized in Tris-HCl (10 mM) pH 8.0 - guanidine (50 mM). Fluorescence in serum and liver extracts was quantified by fluorimetry, as above. Values were normalized per ml of serum or mg of protein. The latter was measured in liver samples using the BCA system (Sigma).
Immunoassays. Western blot and immunoprecipitation were performed as described elsewhere in detail [33]. Densitometric analysis of blots was performed using the Odissey system (Lycor Biosciences, USA). A representative blot is shown from a total of at least three independent experiments. GFAP immunocytochemistry in cultured cells followed previously published procedures [33]. In brief, cultured cells were incubated to block non-specific antibody binding, followed by incubation overnight at 4 °C with anti-GFAP in phosphate buffer (PB) − 1% bovine albumin − 1% Triton X-100 (PBT). After several washes in PB, sections were incubated with an Alexa-coupled secondary antibody (1:1000, Molecular Probes, USA) diluted in PBT. Finally, a 1:500 dilution (in PBS) of DAPI (Hoechst 33342) was added for 3 minutes. Wells were rinsed several times in PB 0.1 N, mounted with 15 µl of gerbatol mounting medium, and allowed to dry. Omission of primary antibody was used as control. Microphotographs were taken in a Leica (Germany) microscope. Plaque load was determined as explained elsewhere in detail [33].
IGF-I in serum and brain was determined using a species-specific ELISA (R&D Systems, USA), as described in detail elsewhere [18]. Murine Aβ (Thermofisher, USA), and murine sAPPα and sAPPβ were determined by ELISA in brain lysates and culture supernatants, respectively, following the manufacturer´s instructions. Blood was collected from the heart after pentobarbital anesthesia and thereafter brains were dissected and frozen at -80ºC until used.
Proximity ligation assays (PLA). Assays were performed as described [34]. Amyloid precursor protein (APP) – IGF-IR interactions were detected in astrocytes and neurons grown on glass coverslips using the Duolink II in situ PLA detection Kit (OLink; Bioscience, Sweden). Cultured cells were fixed in 4% paraformaldehyde/10 min, washed with PBS containing 20 mM glycine to quench the aldehyde groups, permeabilized with the same buffer containing 0.05% Triton X-100 for 5 min, and washed with PBS. After 1 h/37 °C with the blocking solution in a pre-heated humidity chamber, cells were incubated overnight in antibody diluent medium with primary antibodies: mouse monoclonal anti-APP and rabbit polyclonal anti-IGF-I receptor, and processed following the instructions of the supplier using the PLA probes detecting rabbit or mouse antibodies (Duolink II PLA probe anti-Rabbit plus, and Duolink II PLA probe anti-Mouse minus, diluted 1:5 in antibody diluent), and a DAPI-containing mounting medium.
Statistical analysis. Normal distribution tests were carried out in all experiments and a non-parametric Wilcoxon test was applied accordingly. For samples with normal distribution, parametric tests include one-way ANOVA followed by a Bonferroni or t-test. A p < 0.05 was considered significant.
{kind=link}