The browning-sensitive cultivar (36) and browning-resistant cultivar (F) of eggplant showed different degrees of browning with treatment time (Fig. 1). As shown in Table 1, a total of 111.55 GB of clean data were obtained after 18 samples were analyzed and sequenced using a reference transcriptome. The effective data volume of each sample was 5.73–7.04 GB; 93.61%–94.50% of bases had a quality above Q30, and the average GC content was 45.56%. Reads were compared to the reference genome to obtain the genome alignment of each sample, with an alignment rate of 83.50%–95.14%. We also analyzed the distribution of gene expression levels in each sample (Fig. 2). We determined the minimum, the first quartile (25%), the median (50%), the third quartile (75%), and the maximum value of gene expression level for each sample. The symmetry and degree of dispersion of the data distribution were observed, with remarkably high gene expression levels.
Table 1
Summary statistics of clean reads in the transcriptomes of eggplant
|
Sample
|
raw_reads
|
raw_bases
|
clean_reads
|
clean_bases
|
valid_bases
|
Q30
|
GC
|
|
F 15min_1
|
49.75M
|
7.46G
|
48.83M
|
6.63G
|
88.89%
|
94.15%
|
44.08%
|
|
F 15min_2
|
43.77M
|
6.57G
|
42.98M
|
5.84G
|
88.89%
|
93.84%
|
43.55%
|
|
F 15min_3
|
43.15M
|
6.47G
|
42.38M
|
5.77G
|
89.21%
|
94.08%
|
43.46%
|
|
F 2min_1
|
50.03M
|
7.50G
|
49.08M
|
6.54G
|
87.16%
|
93.68%
|
43.82%
|
|
F 2min_2
|
48.79M
|
7.32G
|
47.97M
|
6.49G
|
88.66%
|
94.36%
|
43.43%
|
|
F 2min_3
|
49.04M
|
7.36G
|
48.16M
|
6.52G
|
88.62%
|
94.20%
|
48.59%
|
|
F CK0_1
|
45.73M
|
6.86G
|
44.96M
|
6.11G
|
89.06%
|
94.20%
|
43.50%
|
|
F CK0_2
|
47.44M
|
7.12G
|
46.49M
|
6.20G
|
87.17%
|
93.87%
|
44.36%
|
|
F CK0_3
|
44.35M
|
6.65G
|
43.44M
|
5.75G
|
86.47%
|
93.61%
|
44.04%
|
|
36 15min_1
|
43.53M
|
6.53G
|
42.75M
|
5.81G
|
89.00%
|
94.33%
|
46.06%
|
|
36 15min_2
|
44.28M
|
6.64G
|
43.46M
|
5.95G
|
89.60%
|
94.04%
|
46.80%
|
|
36 15min_3
|
44.47M
|
6.67G
|
43.65M
|
5.89G
|
88.30%
|
94.31%
|
49.09%
|
|
36 2min_1
|
49.59M
|
7.44G
|
48.75M
|
6.57G
|
88.30%
|
94.50%
|
50.06%
|
|
36 2min_2
|
43.62M
|
6.54G
|
42.83M
|
5.73G
|
87.57%
|
94.07%
|
43.78%
|
|
36 2min_3
|
46.19M
|
6.93G
|
45.34M
|
6.05G
|
87.39%
|
94.01%
|
49.84%
|
|
36 CK0_1
|
48.12M
|
7.22G
|
47.28M
|
6.33G
|
87.74%
|
94.03%
|
44.55%
|
|
36 CK0_2
|
52.72M
|
7.91G
|
51.84M
|
7.04G
|
89.07%
|
94.40%
|
44.25%
|
|
36 CK0_3
|
47.98M
|
7.20G
|
47.12M
|
6.33G
|
88.01%
|
94.02%
|
46.79%
|
Gene structure annotation optimization
Currently, new structural features of some genes have not been found yet. We used StringTie [24] software to assemble reads, and then compared the assembled gene with the gene annotation information of the reference sequence. It may be found that the assembled gene extends the 5 'or 3' end of gene annotation, optimizing the starting and ending positions of known genes (Table S1).
SNP/INDEL analysis
Single Nucleotide Polymorphisms (SNP) refer to the variation of a single nucleotide on the genome. The polymorphisms shown in SNP involve only the variation of a single base, which can be caused by the transition (TS) or transversion (TV) of a single base as well as the insertion or deletion of bases. However, the SNP generally do not include the latter two cases, which are represented by INDEL, it is means insert and delete.
The differences between the eggplant varieties used in the experiment and the eggplant varieties with the reference genome, and the presence of RNA editing in the samples treated differently, may lead to SNP mutation sites [25]. Tools such as Samtools[26], BedTools [27] were used to predict SNP and INDEL sites in samples. The number of SNP sites found in each sample were 187982 (F 15min), 185271 (F 2min), 177684 (F CK0), 158720 (36 15min), 138662 (36 2min) and 190694 (36 CK0), mainly distributed in exon region, intron region, intergenic region and upstream and downstream regions of genes (Table S2). The number of INDEL sites found in each sample were 11478 (F 15min)、11905 (F 2min)、11503 (F CK0)、158720 (36 15min)、9275 (36 2min)、12472 (36 CK0),mainly distributed in upstream and downstream regions (Table S2). In addition, we divided the number of SNP/INDEL loci of each gene by the length of the gene to obtain the density value of SNP loci of each gene, counted the density value of SNP/INDEL loci of all genes and made the density distribution map (Fig. S1).
SNP sites can be classified into transition and transversion depending on how the base is replaced. Among the samples in different treatment groups, the highest frequency of transversion was SNP(A/T), and the number of SNP sites was 10438 (F 15min), 10181 (F 2min), 9814 (F CK0), 8489 (36 15min), 7513 (36 2min) and 10192 (36 CK0), respectively (Table S3). The highest frequency of transition was SNP(A/G), and the number of SNP sites was 37700 (F 15min), 37412 (F 2min), 35932 (F CK0), 32396 (36 15min), 28518 (36 2min) and 38527 (36 CK0), respectively. The ratios of Ts/Tv were 2.017(F 15min), 2.042 (F 2min), 2.025 (F CK0), 2.036 (36 15min), 2.039 (36 2min) and 2.040 (36 CK0), respectively (Table S3).
Identification and classification of DEGs
Identification of DEGs between the two eggplant cultivars was one of the aims of the present study. To achieve this goal, we compared the transcriptional profiles of cultivars 36 and F, which were used as a test and a control for screening DEGs, respectively. According to the standard criteria (fold change > 2 and p-value < 0.05), we analyzed the difference in the expression of the same gene between treatment pairs. For cultivar 36, we identified 4095 DEGs between treatment 1 and the control (1971 and 2124 up- and down-regulated genes, respectively), 2212 DEGs between treatment 2 and the control (857 and 1355 up- and down-regulated genes, respectively), and 1957 DEGs between treatment 2 and treatment 1 (996 and 961 up- and down-regulated genes, respectively). For cultivar F, we identified 20 DEGs between treatment 1 and the control (11 and 9 up- and down-regulated genes, respectively), 1313 DEGs between treatment 2 and the control (1188 and 125 up- and down-regulated genes, respectively), and 1087 DEGs between treatment 2 and treatment 1 (1014 and 73 up- and down-regulated genes, respectively). Furthermore, there were 2299 DEGs between the control of cultivar 36 and that of F (1363 and 936 up- and down-regulated genes, respectively), 2810 DEGs between treatment 1 of cultivar 36 and that of F (1633 and 1177 up- and down-regulated genes, respectively), and 3820 DEGs between treatment 2 of cultivar 36 and that of F (1336 and 2484 up- and down-regulated genes, respectively) (Fig. 3). The DEGs were then subjected to GO and KEGG pathway enrichment analyses.
Functional annotation and classification of DEGs
To determine the function of DEGs, GO and KEGG pathway analyses were carried out. For cultivar 36, the treatment pair with the most DEGs was the treatment 1/control pair. In this treatment pair, GO functional enrichment analysis was carried out (Fig. 4a). “Metabolic process,” “cell,” and “binding” were the dominant terms in the “biological process,” “cellular component,” and “molecular function” categories, respectively. We also identified a relatively large number of genes associated with “catalytic activity,” “antioxidant activity,” “biological regulation,” “response to stimulus,” “single-organism process,” and “cellular process,” with only a few genes related to “cell killing,” “nucleoid,” and “protein binding transcription factor activity.” As shown in Fig. 4b and c, GO functional enrichment analysis of the treatment pairs F treatment 2/F control and 36 treatment 2/F treatment 2 showed similar results as the 36 treatment 1/36 control treatment pair.
We used KEGG pathway analysis to determine the biological functions of DEGs. Pathway entries with a number of corresponding DEGs greater than 2 were screened and sorted by the -log10p value of each entry. The top 20 pathways were retained. In the 36 treatment 1/36 control pair (Fig. 5a), the DEGs were mostly involved in phenylpropanoid biosynthesis, flavonoid biosynthesis, tyrosine metabolism, pentose and glucuronate interconversions, and starch and sucrose metabolism. In the F treatment 2/F control pair (Fig. 5b), the DEGs were mostly involved in the AMPK signaling pathway, metabolism of xenobiotics by cytochrome P450, drug metabolism - cytochrome P450, plant hormone signal transduction, and starch and sucrose metabolism. However, in the 36 treatment 2/F treatment 2 pair (Fig. 5c), the DEGs were mostly involved in phenylpropanoid biosynthesis, flavonoid biosynthesis, glyoxylate and dicarboxylate metabolism, stilbenoid, diarylheptanoid and gingerol biosynthesis, and tyrosine metabolism.
Identification of potential genes associated with enzymatic browning
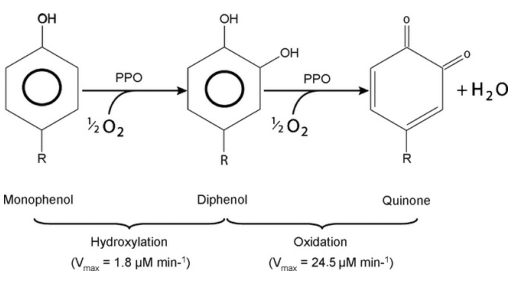
Enzymatic browning is one of the leading causes of tissue browning in fresh-cut fruits and vegetables. We analyzed the expression of the key enzymatic browning enzymes, and found that four PPO-encoding genes (Sme2.5_25992.1_g00001.1、Sme2.5_01441.1_g00006.1、Sme2.5_06843.1_g00001.1、Sme2.5_33651.1_g00001.1), two POD-encoding genes (Sme2.5_00423.1_g00003.1, Sme2.5_00745.1_g00004.1), one CAT-encoding gene (Sme2.5_01411.1_g00008.1), one PAL-encoding gene (Sme2.5_16832.1_g00001.1), and one CHS1B-encoding gene (Sme2.5_13923.1_g00001.1) had higher transcript levels in cultivar 36 than in cultivar F. Three genes had higher transcription levels in cultivar F compared with cultivar 36, one COMT-encoding gene (Sme2.5_29908.1_g00001.1), one GST-encoding gene (Sme2.5_09086.1_g00002.1), and one L-ascorbate oxidase-encoding gene (Sme2.5_05133.1_g00002.1). In addition, we identified two transcription factors (Sme2.5_00574.1_g00007.1, Sme2.5_07339.1_g00001.1) with differential expression in the two species; these proteins belong to the WRKY transcription factor gene superfamily, which affects different physiological responses and metabolic pathways (Fig. 6, Table S4).
Validation of differentially expressed genes with quantitative real-time PCR
Heat maps for the 12 browning-related genes and two WRKY transcription factor indicated that the expression levels varied with both cultivar and treatment (Fig. 6). In order to confirm the expression level of the 12 genes and two WRKY transcription factors, qRT-PCR was carried out. The design list of primers is shown in Table S5. The quantitative results are shown in Fig. 7. The results of qRT-PCR were consistent with the analysis of the transcriptome, confirming the reliability of the RNA-seq data.
{kind=link}