Relevant information of all commercially available reagents was provided in Additional file 1.
Investigational product
The antigenused in this study is a recombinant fusion protein, representative of human VEGF isoform 121 (P64KhVEGFKDR-) [18]. The lyophilized antigen was produced in vials of 400 µg by the Development Unit of Center for Genetic Engineering and Biotechnology (CIGB, Havana, Cuba). The antigen was mixed with the adjuvant VSSP, very small sized particles obtained from the Neisseria meningitides outer membrane, supplied by the Center of Molecular Immunology (CIM, Havana, Cuba). Both, antigen and VSSP were produced under GMP conditions. At the moment of vaccination, one antigen vial was dissolved in pre-calculated amounts of injection water, and the required amount was mixed with the established quantity of VSSP (200 µg), up to a final volume never exceeding 1mL per injection dose.
Compassionate use program (CUP)
This CUP is a study of the CIGB–247 vaccine, and was conducted in accordance with the Regulation 63–2012 emitted by the Cuban Regulatory Authority (CECMED) [26]. This investigation was also performed in compliance with the ethical guidelines of the Declaration of Helsinki. All physicians who wanted to include patients in the CUP were required to contact the main coordinator of the study via e-mail ([email protected]), attaching the following documents: (1) formal request letter asking the compassionate use of CIGB–247 and indicating that the patient received all available onco-specific therapies without response; (2) summary of the patient´s medical record; (3) approval letter by institutional ethics committee. After that, the main coordinator send to the physician the informed consent form and other relevant information about the vaccine candidate (list of references, management of adverse events, safety profile). To participate in the CUP, all patients had to sign the informed consent form, after which the physician submitted the document through e-mail and send the following information: patient´s full name, age, sex, type of cancer (solid tumor or hematologic malignancy), histopathological diagnosis, and the presence or not of metastases. In case of child participant, it was mandatory the assent from children (whenever possible) and a written informed consent from both parents or legal guardians. Once approved by the main coordinator, the patient was assigned a code, and the procedure of the vaccine preparation, immunization schedule and times of blood sampling were send to the physician by the main coordinator. Subsequently, the Center for Genetic Engineering and Biotechnology delivered the antigen and the adjuvant to the hospital for patient treatment. The physician was only obligated to report adverse events, probably or definitively related to the vaccine. Also, it was not mandatory the acquisition of blood samples.
Patient inclusion criteria and immunization protocol
CUP in Cuba included subjects of any sex and age, diagnosed with solid tumor or hematologic malignancy in advanced stages, with non-measurable or measurable lesions, metastasis free patients or individuals with metastatic disease of any localization. There was no restriction for Eastern Cooperative Oncology Group (ECOG) performance status, for chronic un-compensated diseases, autoimmune or immune suppressing diseases. Patients receiving immune modulator drugs, chemotherapy or biological therapies including active or passive immunotherapy were also recruited. Subjects with allergies to vaccine components, pregnancy or breast feeding, and evident mental incapacity to understand the information, deliver the consent, and act in consequence during the study were excluded.
One hundred fifty three patients were immunized with 400 µg of antigen in combination with 200 µg of the adjuvant VSSP, which represents the highest antigen dose that at that point of the initiation of this program had been found to be safe. All vaccinations were administered subcutaneously as a single site dose. Each patient received eight weekly vaccinations followed by a re-immunization on week twelve (induction phase). Four weeks after the ninth vaccination (week sixteen), re-immunizations were done once every four weeks, until death, intolerance, marked disease progression or patient´s withdrawal of consent (re-immunization phase).
Human blood samples
Blood samples were obtained from 41 of the 153 vaccinated patients. Venous blood samples were collected using a blood collection set with pre-attached holder and taken into a serum separator tube for serum analyses. Serum samples were obtained as has been previously described [27, 28], and were immediately stored at –20ºC or –70ºC until use.
Blood samples were taken before initial vaccination (week 0 or pre-vaccination sample) and during the induction phase or re-immunization phase. For investigations conducted during both phases, blood samples were taken at different time points, depending on patient availability.
Biotinylation of monoclonal antibody Bevacizumab and VEGF binding testing
To develop a competitive ELISA that measures the inhibition of the binding between VEGF and Bevacizumab, this monoclonal antibody was biotinylated. A Bevacizumab solution of 3.68 mg/mL was obtained in labelling buffer (0.1M NaHCO3, 0.1M NaCl pH 8.5) by exchange chromatography. Biotin N-hydroxysuccinimide ester was added to Bevacizumab solution at a ratio of 0.1 mg of biotin per mg of antibody. The reaction mixture was incubated with stirring during 4h at room temperature. Free biotin was removed and antibody was exchanged into PBS by gel filtration. VEGF binding curves for Bevacizumab and biotinylated Bevacizumab yielded similar half maximal effective concentration (EC50), indicating that conjugation did not affect the antigen- binding site (Additional file 2).
ELISAs reagents
Human VEGF isoform 121 (hVEGF CHO) and human VEGF-C (hVEGF-C CHO) were produced in CHO cells [29]. Plasmid construction and cell line development for hVEGF-C CHO are described in Additional file 3. HRP-conjugated goat anti-human IgG (Fc γ fragment specific) antibody was used at 80 ng/mL for detecting human serum IgG. Biotinylated goat antibodies specific for human VEGFR2 or human VEGFR1 were used at 0.1 µg/mL for detecting VEGF/VEGFR2 or VEGF/VEGFR1 bindings respectively. Streptavidin-peroxidase conjugate was used 1/30 000 or 1/35 000 dilution. Human serum, positive (PCS) and negative (NCS) for VEGF-specific IgG antibodies, have been previously used as assay controls [29].
ELISA for specific anti-human VEGF IgG, IgM, IgA and IgE antibodies
The levels of human IgG, IgM, IgA and IgE antibodies against VEGF were measured as described previously [22, 29]. Briefly, wells were coated with hVEGFCHO during overnight incubation at 4ºC. Following blocking step, the wells were incubated with samples and IgG, IgM, IgA or IgE antibodies were detected with HRP-conjugated goat anti-human IgG antibody, biotinylated goat anti-human IgM antibody, biotinylated anti-human IgA monoclonal antibody or biotinylated anti-human IgE monoclonal antibodies, respectively. For biotinylated conjugates the detection system consisted of streptavidin-conjugated HRP. Plates were developed by using H2O2 as substrate and TMB as chromogen.
IgG antibody titer was estimated as previously described [29]. The procedure was similar for IgM, IgA and IgE with the difference that the interpolated value on “x” axis was determined by adding five standard deviations to the duplicated mean of the blank optical density.
Titer ratio and “VEGF-specific antibody titer” were calculated as follow:
[Due to technical limitations, this equation is only available as a download in the supplemental files section.] (A)
Specific antibody titer = Post vaccination titer-Pre vaccination titer (B)
To declare a given sample taken during vaccination to be positive for VEGF-specific IgG, IgM, IgA, or IgE antibodies, the obtained “titer ratio” must be ≥2 (formula A). In the particular case of IgG antibodies, additionally to the criterion depicted above, for a sample to be considered positive, it has also to comply with a value of “specific antibody titer” ≥1/100 (formula B).
The term seroconversion is only used in this paper for IgG antibodies and refers to a patient that has shown two or more samples positive for VEGF-specific antibodies during re-immunization phase (seroconverted patient) [21].
IgG subclasses assays
VEGF-specific IgG1, IgG2, IgG3, and IgG4 antibodies were determined as previously described [22]. Briefly, microtiter plates were coated with hVEGF CHO during overnight incubation at 4ºC. Following blocking step, sera were added and antigen-specific IgG1, IgG2, IgG3, and IgG4 antibodies were detected using biotinylated mouse monoclonal anti-human subclass-specific antibodies.
To declare a given serum sample taken during vaccination as “non-detectable” for VEGF-specific IgG1, IgG2, IgG3, or IgG4 antibodies, “specific antibody titer” must be < 1/10. Values ≥ 1/10 make samples to be classified as “detectable”. For each patient, the IgG subclass classified as “detectable” with the highest “specific antibody titer” was declared as “predominant” [22].
Competitive ELISA evaluating the blockade of the binding between VEGF and its receptors
Competitive ELISA has been previously described in details by Sánchez et al. [22, 29]. Briefly, plates were coated with hVEGF CHO during overnight incubation at 4ºC. Following blocking step, sample was added and incubated for 1 h at 37 ºC. Then, 100 µL of 25 ng/mL of VEGFR2-Fc or 125 ng/mL of VEGFR1-Fc were added to the wells (12.5 and 62.5ng/mL final concentration respectively) and additionally incubated for 45 min at 37 ºC. After washes, wells were incubated with biotinylated anti-human VEGFR2 or VEGFR1 antibodies, the latter followed by streptavidin-peroxidase conjugate.
Maximum bindings of VEGFR2 or VEGFR1 were obtained from wells incubated with dilution buffer (instead of sample) and VEGF receptors/Fcγ chimeras (VEGFR2-Fc or VEGFR1-Fc). The inhibition caused by a given sample on VEGF/VEGFR2 or VEGF/VEGFR1 interactions was expressed as percentage, according to the following formula:
[Due to technical limitations, this equation is only available as a download in the supplemental files section.] (C) (D)
A given sample was considered positive for blocking activity when the value resulting from this ratio was ≥2 (formula D). Patients showing at least one serum sample with neutralizing anti-VEGF antibodies during induction phase or re-immunization phase were considered with a positive blocking activity on the VEGF/VEGFR1 or VEGF/VEGFR2 bindings [21].
Competitive ELISA evaluating the blockade of the interaction between VEGF and Bevacizumab
Plates were coated with hVEGF CHO (1µg/mL in PBS, 100µL/well, overnight incubation at 4ºC). After three washes with 0.1% Tween 20 in PBS, the plates were blocked with 2.5% goat serum (v/v), 2% skim milk (m/v), 0.05% Tween 20 (v/v) in PBS (250µL/well, 1h at 37ºC). After a washing step, test sample or dilution buffer were added (100 µL/well, 1h at 37ºC). Then, 100 µL/well of 7.6 ng/mL of biotinylated Bevacizumab antibody were added to the wells (3.8 ng/mL final concentration and diluted in blocking buffer) and additionally incubated for 1h at 37 ºC. The maximum binding of Bevacizumab was obtained from incubated wells with dilution buffer (instead of sample) and biotinylated Bevacizumab. After washes, wells were incubated streptavidin-peroxidase conjugate (diluted 1:30 000 in 1% BSA/PBS, 100 µL/well, 45 min at 37ºC). After washes, the subsequent steps of the reaction were developed as described in previous sub-sections.
Each plate included “blank” wells that were developed in parallel and did not receive neither test samples nor biotinylated bevacizumab, only dilution buffer. The other ELISA steps (antigen coating, adding blocking buffer, incubating with biotinylated antibody and streptavidin-peroxidase conjugate, adding substrate and stopping buffer) were performed as those for other wells.
The inhibition caused by a given sample on VEGF/Bevacizumab interaction was expressed as percentage, according to formula C. The final concentration of 3.8 ng/mL corresponds to half maximal effective concentration (EC50), and this value was obtained from four independent experiments (Additional file 4).
ELISA for detecting human IgG antibodies specific to VEGF-C and VEGF-D
Two strategies were used for detecting human IgG antibodies specific to VEGF-C and VEGF-D. For the first one, wells were coated with a monoclonal antibody specific to myc-tagged proteins (10 µg/mL in PBS, 100µL/well, overnight incubation at 4ºC). Following a washing step (0.12% Tween 20 v/v) and a blocking step (2.5% goat serum v/v, 2% skim milk m/v, 0.05% Tween 20 v/v in PBS, 250µL/well, 1h at 37ºC), the wells were incubated with hVEGF-C CHO or hVEGF CHO (5 µg/mL in blocking buffer, 100µL/well, 1h at 37ºC). Plates were washed, and test samples were added (diluted in blocking buffer, 100µL/well, 1h at 37ºC). Specific IgG antibodies were detected with HRP-conjugated goat anti-human IgG antibody (diluted in 2% skim milk v/v in PBS, 100µL/well, 1h at 37ºC). Plates were developed by using H2O2 as substrate and TMB as chromogen (100 µL/well, 10 minutes at room temperature). The reaction was stopped by the addition of 2.0 N H2SO4 (50 μl/well), and the absorbance was measured at 450nm.
For the second strategy, VEGF-C or VEGF-D (2.5 µg/mL in PBS, 100µL/well, overnight incubation at 4ºC) were added to Histidine-select nickel coated high sensitivity multiwell plates. After a washing step (0.05% Tween 20 v/v in PBS), test samples were added (diluted in 0.05% skim milk v/v, 0.05% Tween 20 in PBS, 100µL/well, 2h at 37ºC), and specific IgG antibodies were detected with HRP-conjugated goat anti-human IgG antibody (diluted in 0.05% skim milk v/v, 0.05% Tween 20 in PBS). The subsequent steps of the reaction were developed as previously described. Recombinant human VEGF receptor 3/Fcγ chimera was used as assay positive control.
IgG fraction purification
Post-vaccination sera from different patients and positive for VEGF-specific IgG antibody titer were pooled, and IgG from serum was purified by protein A. Lipoproteins were removed by adding solid PVP to the serum to a final concentration of 3% (w/v). After 4h at 4ºC, sample was centrifuged at 15 700 g for 30 min at 4ºC. Supernatant was removed and exchanged into 0.02 M sodium phosphate buffer pH 7 (binding buffer) using a desalting column. After centrifugation, supernatant was mixed to a pre-equilibrated protein A sepharose media, and later incubated during 14–16h at 4ºC with stirring. The gel bed was added to an empty column, and the excess fluid was allowed to drain via gravity. The gel bed was washed with binding buffer, and the IgG fraction was eluted 0.1 M glycine buffer pH 2.7. The IgG fraction was collected into a neutralization buffer (1M Tris-HCl pH 9). The eluate was immediately exchanged into PBS, concentrated between 10–20 mg/mL of IgG, and the final sample was stored at –70ºC until use (IgG comp). A purified human IgG isolated from pooled normal human serum (IgG neg) was used as assay negative control.
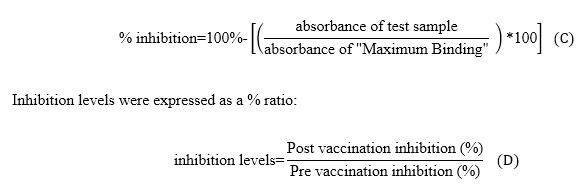{kind=link}
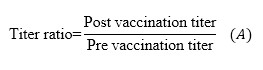{kind=link}