Sample collection and extraction of nucleic acids
Field surveys and sampling were carried out between January and May 2018 in four major tomato growing regions in Kenya, with different agro-ecological and climatic conditions (Figure 1a). Tomato fields were randomly selected based on crop availability, with 30 plants randomly assessed per field. From each field, young trifoliate leaf samples (n=5) were obtained only from plants showing symptoms such as chlorosis, reduced leaf size, upward leaf curling, stunting and flower abscission (Figure 1b). A total of 240 leaf samples were collected from 48 fields, carried in paper bags and stored at -80 °C till further analysis. Samples from each field were pooled prior to DNA extraction.
Extraction of total genomic DNA was performed as described [26]. Briefly, about 150 mg of leaf tissues were homogenized using a mortar and pestle with 1.5 ml of pre-warmed extraction buffer (2% cetyl trimethyl ammonium bromide w/v; 100 mM Tris-HCl, 1.4 M NaCl, 20 mM EDTA, pH 8.0 + 50 mg PVP + 0.2% v/v β –mercaptoethanol added just before use). The samples were transferred into 1.5 ml microtubes and incubated at 65ºC for 30 mins while mixing at 10 mins interval. The tubes were centrifuged at 10,000 rpm for 5 secs and supernatants (750 µl) were transferred into fresh microtubes. Chloroform and isoamyl alcohol (750 µl) in the ratio 24:1 was added to the tubes, mixed and centrifuged at 10,000 rpm for 15 mins. The aqueous layers were transferred into new microtubes and ice cold isopropanol (300 µl) were added and mixed by inverting the tube slowly. Tubes were incubated overnight at -20ºC and the nucleic acids were then pelleted by centrifugation at 10,000 rpm for 15 mins. The supernatants were discarded, pellets washed with 500 µl of 70% (v/v) ethanol and dried at room temperature. These were dissolved in 100 µL of Tris-EDTA buffer (10mM Tris-HCl [pH 8.0] + 1 mM EDTA), incubated at 37ºC for 30 mins and stored at -20ºC. A Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA) was used to determine the quality of the nucleic acids.
Library preparation and sequencing
The genomic DNA were quantified using a QubitTM fluorometer (Thermo Fisher Scientific, MA, USA) and normalized to 2.5 ng/µl and used for library preparation. Libraries were prepared using Nextera DNA library preparation kit (Illumina, CA, USA) according to the manufacturer’s instructions. Briefly, enzymatic fragmentation was carried out on normalized genomic DNA samples (20 µl) via addition of TD buffer (25 µl) and TDE (5 µl). Mixtures were centrifuged (Hettich Centrifugen, D–78532, Germany) at 14,000 rpm at 20 oC for 1 min and transferred into microtubes. Tagmentation was carried out in a pre-programmed thermocycler at 55 oC lid and 55 oC incubation temperature, while holding at 10 oC. The tagmented DNA was barcoded using indexed adapters then cleaned with AMPure XP magnetic beads (Beckman Coulter, Inc. Indianapolis, IN) to remove shorter DNA fragments and other impurities. Library quality was confirmed with the Agilent Tape Station 2200 System (Agilent Technologies, Santa Clara, CA). All the 48 libraries were quantified using the QubitTM fluorometer (Thermo Fisher Scientific Inc., Waltham, MA). The indexed DNA libraries of 48 biological samples were each normalized to a concentration of 4 nm before being pooled together. High-throughput sequencing was performed on an Illumina MiSeq System using 2 × 251 v2 kit and 12 pM of 1% PhiX v3 spike to create paired-end reads. Sequencing was performed at the facility of the Biosciences Eastern and Central Africa International Livestock Research Institute (BecA-ILRI) Hub, Nairobi, Kenya.
Sequence processing and assembly
After sequencing, quality control of fastq paired end reads was performed using FastP v.0.20.0 [27] to remove adapters, poly-N sequences (≥15%) and filter off low quality reads. High-quality reads were then mapped to the tomato genome (GenBank RefSeq accession number GCA_000188115.3) using Bowtie v.2.3.4.3 [28] under default parameters. Unmapped reads were assembled into contigs de novo using MEGAHIT v.1.1.3 [29] with default settings and those representing ssDNA sequences were verified using Kaiju virus database [30]. The sequences were then subjected to BLASTN 2.9.0+ [31] to determine similarity match and virus identification (Additional File 1: Figure S1). Protein prediction of ORFs was determined using ORF Finder (http://www.ncbi.nlm.nih.gov/projects/gorf).
Sequence validation through Polymerase chain reaction (PCR) and Sanger sequencing
The assembled begomovirus genomes were validated using a polymerase chain reaction (PCR) step followed by Sanger sequencing of the amplified products. The Illumina assembled virus sequences were aligned together using ClustalW multiple sequence alignment program with default parameters as implemented in BioEdit v.7.2.3 [32]. A consensus sequence was obtained and used to design PCR primers ToLCV-Forward (5’-ATTGGCGATTTCCCAGGTATAG-3’) and ToLCV-Reverse (5’-ACAATGTGGGCTAGGTCATTAG-3’) using the Primer Express v3.0 software (Applied Biosystems, USA). Secondary structures, complementarity and dimer effects of the primers were also checked using the multiple primer analyzer software (Thermo Fisher Scientific, MA, USA). Using PCR, these were tested on the genomic DNA from which the complete begomovirus genomes had been obtained via Illumina sequencing. The PCR products were ethanol-purified and quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA) to determine purity levels. Amplicons were sequenced at Macrogen Europe and manually assembled using BioEdit. Consensus sequences were verified using BLASTN 2.9.0+ and comparisons were made with the complete begomovirus sequences assembled from Illumina reads.
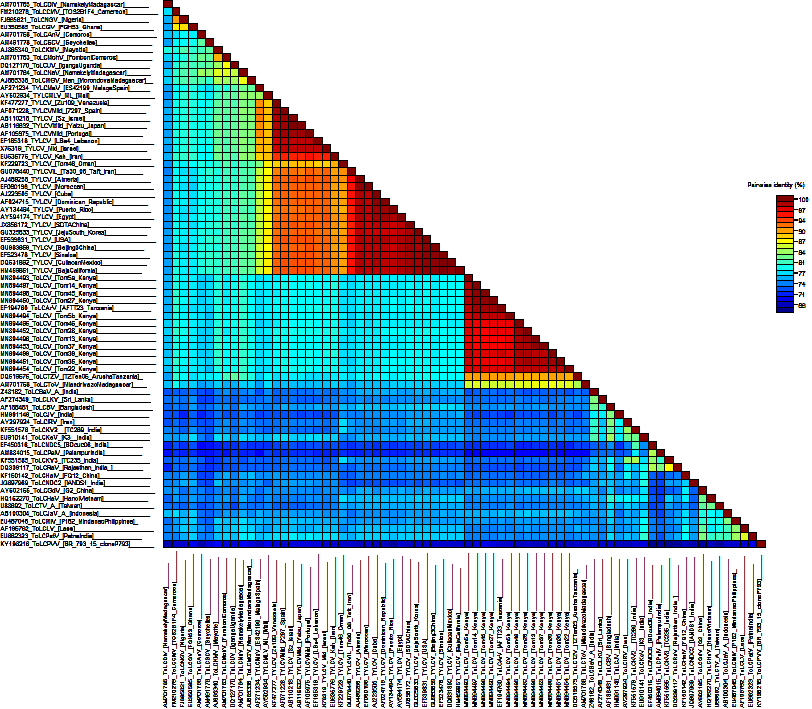
Sequence alignment, distance matrix and evidence of recombination
Complete sequences of monopartite begomoviruses found in tomato were retrieved from GenBank (Additional File 2: Table S1) and aligned with full virus contigs using ClustalW in BioEdit. Deduced amino acids from the ToLCV genomes were compared with GenBank isolates while sequence pairwise identities were performed using SDT v1.2 [33] with pairwise gap deletions. A scan for recombination signatures were performed on each protein-coding sequence data using the single breakpoint scanning (SBP) and genetic algorithm recombination detection (GARD) methods [34]. These two methods were implemented by the Datamonkey software [35]. Potential recombination events were further investigated using the default settings of the seven detection algorithms within RDP v 4.13 [36]. Putative recombination events, potential recombinants, and their parental sequences were deemed acceptable only when signals were identified by at least four detection methods, with strong levels of significance (P≤0.05).
Phylogeny, genetic diversity and population genetic analysis
A phylogenetic tree was constructed using the maximum likelihood method based on Jukes-Cantor model in MEGA v.6.06 [37]. Bootstrap replicate values were set at 1,000 while a strain of Tomato leaf curl purple vein virus (KY196216) was selected as an outgroup. Genetic structure and diversity within ToLCV populations in Kenya were investigated to understand potential evolutionary dynamics that produce variations. Population structure parameters estimated included; average nucleotide diversity (π), haplotype diversity (Hd), number of polymorphic or segregating sites (S), the statistic estimate of population mutation based on the number of segregating sites (θ-W), total number of mutations (Eta), the average number of nucleotide differences between sequences (k) and the statistic estimate of population mutation based on the total number of mutations (θ-Eta). These were estimated using complete genome and protein coding sequences in DnaSP v5.10.01 [38].
The possible occurrences of selection pressure on individual genes and sites within the ToLCV populations were obtained using the single-likelihood ancestor counting (SLAC) method [39] in the HyPhy package [40] as implemented on the Datamonkey software [35] at http://www.datamonkey.org. The ratio of average number of nucleotide differences between the sequences per nonsynonymous site (dN) to the average number of nucleotide differences between the sequences per synonymous site (dS) were calculated as an indicator of natural selection. These were used to estimate the occurrence of positive and negative selection at typical begomovirus amino acid ORF sites: the movement protein (MP) or V1 protein, coat protein (CP) or V2 protein, replication protein (Rep) or C1 protein, transcription activator protein (TrAP) or C2 protein, Rep enhancer protein (REn) or C3 protein and the C4 protein. Depending on the dN/dS values, the selection pressure was considered negative or purifying (dN/dS < 1), neutral (dN/dS = 1), or diversifying or positive (dN/dS > 1) for data sets of each coding region. The DNAsp v5.10.01 was used to calculate the Tajima's D, Fu and Li's F* and D*, and Fu's Fs to determine the deviation of ToLCV populations from neutrality assuming a constant population size, with zero recombination and migration [41]. A negative Tajima’s D statistic indicates superfluous low-frequency polymorphism triggered by background selection, genetic hitchhiking, or population expansions [42]. Conversely, positive values of Tajima’s D statistic suggest minimal levels of low and high frequency polymorphisms, indicating a reduction in population size and/or balancing selection.
{kind=link}
{kind=link}