2.1 Materials and reagents
Low-temperature defatted flaxseed proteins were obtained from Inner Mongolia Fengjimiao Agricultural Products Technology Development Co., Ltd. Caco-2 and HepG2 cells were purchased from the Institute of Biochemistry and Cell Biology (CAS; Shanghai, China). Protease M was obtained from Nippon Amano Enzyme Co., Ltd., Japan. The following materials were used: an AL204 electronic balance (Mettler Toledo Co., Ltd., Switzerland), an 8200 stirred ultrafiltration cell (Millipore Co., Ltd., USA), and a FreeZone® 4.5 desktop freeze dryer (American Labconco Corporation, USA). Electrophoresis, a transfer tank (Bio-Rad, USA), and a MultiSkan3 microplate reader (Thermo Fisher Scientific, USA) were also utilized. Ultimate 3000 HPLC-ESI-Orbitrap MS with Protect-1FD Ultra-clean workbench (Agilent Co. Ltd, USA). CO2 incubator (Thermo Fisher Scientific, USA), and an inverted microscope (Nikon Co., Ltd., Japan). MEM basic medium, bovine albumin, and antibiotics were procured from Sigma Co., Ltd., USA. Millicell electrical resistance system (ERS) and electrical resistance meter were acquired from Thermo Fisher Scientific, USA. Anti-β-actin, NPC1L1, ABCG5/G8 (1:1000, rabbit, Abcam Inc., UK), cell lysate, Western blot kit (NJJC, Nanjing, China). Transcriptor First Strand cDNA Synthesis Kit (Roche, Berlin, Germany). SYBR® Premix EX Taq II Kit (Takara, Shiga, Japan). Thermal Cycler Mx3000P QPCR System (Agilent, CA, USA)
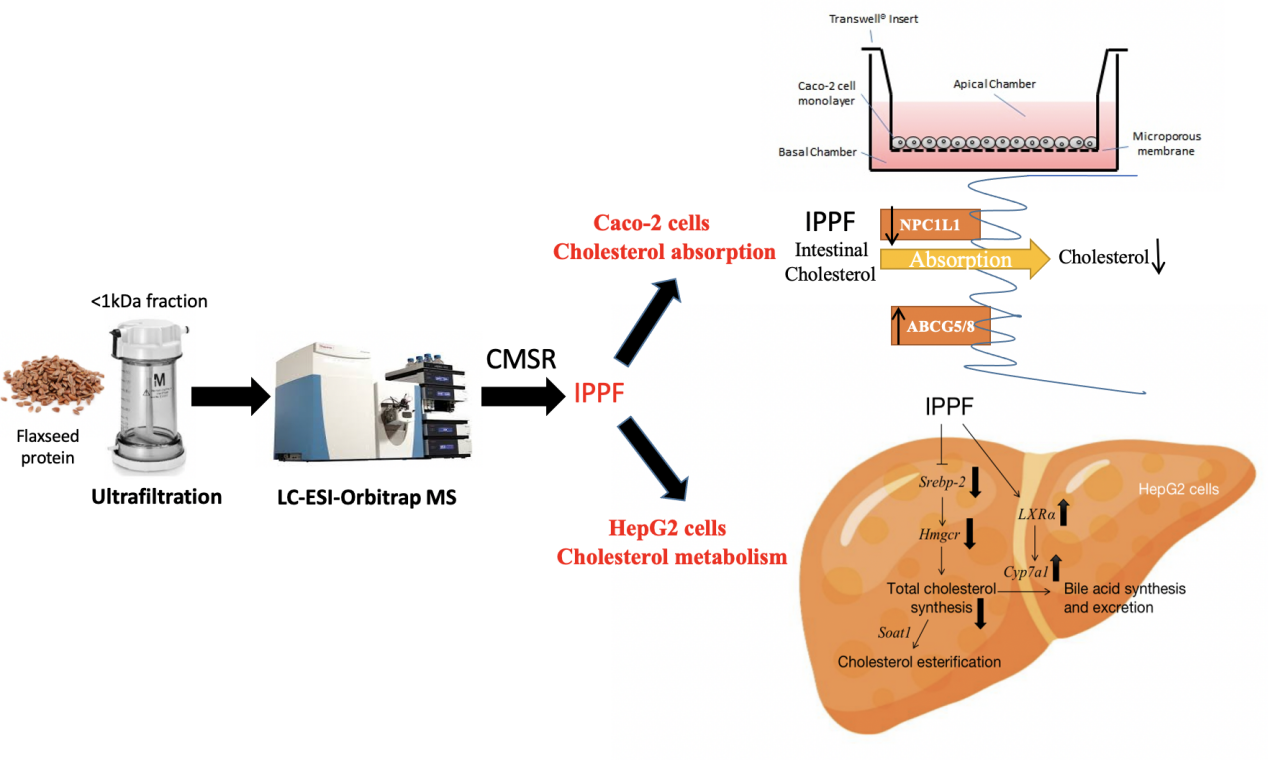
2.2 Preparation of flaxseed protein hydrolysates
First, 1 g of protein was weighed in a beaker and added with 50 mL of distilled water. pH was adjusted to 3.3 by using 1 mol/L HCl. The solution was then placed on a constant-temperature magnetic stirrer and added with 0.01 g of protease M (optimum pH = 3.3, optimum temperature = 50°C, E/S ratio = 1:100) when the temperature reached 50°C [10]. The temperature was maintained at 50°C, and enzymatic hydrolysis was conducted for 1 h. The enzyme was degraded by incubating the solution in a boiling water bath for 15 min. The solution was cooled to room temperature and centrifuged at 1792 r/min for 20 min. Afterward, pH was adjusted to 7 by adding 1 mol/L NaOH, and then the supernatant was freeze-dried.
2.3 Cholesterol micelle solubility inhibition rate of different FP fractions
FPHs were separated using an ultrafiltration with molecular weight of 1, 3, 5, and 10 kDa. Five types of ultrafiltration components of ≤ 1, 1–3, 3–5, 5–10, and ≥ 10 kDa were separated via ultrafiltration. Cholesterol micelles were prepared in accordance with the method of Zhang et al [11], with some modifications. Subsequently, 5 mg of different FPH fractions was dissolved in 1 mL of micelle solution. The solution was shaken at 37°C for 24 h and centrifuged at 15,000 r/min for 60 min. The supernatant fractions (25 µL) were collected, and cholesterol concentrations were determined using a cholesterol assay kit (NJJC, Nanjing, China) by measuring the absorbance at 510 nm. Finally, CMSR was calculated.
2.4 LC-ESI-Orbitrap MS analysis of FP5
The amino acid sequences of FPH5 were determined via HPLC-ESI-Orbitrap MS.
For the HPLC conditions, the sample was reconstituted in 0.1% aqueous formic acid, filtered through a 0.45 µm microporous membrane, and separated by a reverse-phase C18 column. The mobile phase A comprised 0.1% aqueous formic acid, whereas the mobile phase B comprised 0.1% formic acid in acetonitrile. The elution gradients with respect to mobile phase B were 0–5 min, 6–9%; 5–20 min, 9–14%; 20–50 min, 14–30%; 50–58 min, 30–40%; and 58–60 min, 40–95%. The total elution time was 60 min, and the column temperature was 30°C [13]. The detection wavelength was 220 nm, the injection volume was 10 µL, and the flow rate was 300 nL/min. The eluent was directly placed in the mass spectrometer system for analysis.
For ESI-Orbitrap MS conditions, the detection mode of ESI was in positive ion source, the capillary temperature was 250 ℃, the electrospray voltage was 2,100 V, and the m/z sweep range was 100–1,800.
The original files of the mass spectrometer were processed with the supporting commercial software Maxquant (1.6.2.10). The obtained peptide sequences were compared with the amino acid sequences of the protein database (UniProtKB) to obtain the matched amino acid sequences.
2.5 Solid-phase peptide synthesis and CMSR of synthesized peptides
The peptide amino acid sequences from FPH5 were synthesized by ChinaPeptides Co. (Shanghai, China) by using a solid-phase peptide synthesis (SSPS) procedure. The molecular masses and purities (> 97%) of the peptides were determined using an HPLC-MS system with a C18 column. A solution that consisted of 60% CH3OH, 39.9% H2O, and 0.1% HCOOH was used as the mobile phase. The flow rate was set to 0.5 mL/min, and the detection range was m/z 100–1800. The CMSR of the synthesized peptides was then evaluated.
2.6 Preparation of IPPF antibody
Immunization protocol: On the 1st day, the treated New Zealand rabbits were disinfected with alcohol, and 1 mL of antigen was added to 1 mL of Freund’s complete adjuvant. The mixture was injected at multiple points (at least eight points) under the skin on the back of the neck. On the 15th day, the first booster immunization was performed, and 1 mL of antigen was added to 1 mL of Freund’s incomplete adjuvant for emulsification then was injected at multiple sites under the skin on the back of the neck. On the 29th day, the second booster immunization was performed. On the 43rd day, the third booster immunization was performed. On the 53rd day, blood was taken from the carotid artery, and the rabbits were sacrificed. The rabbits’ blood was placed overnight at 4°C, centrifuged at 10000 r/min for 30 min at 4°C, and the supernatant (serum, IPPF antibody) was collected.
Preparation of primary and secondary antibodies: After the IPPF antibody was purified, the antibody titer was determined by ELISA. The titer reached the target of about 1:1250. On the 63rd day, blood was collected via cardiac puncture. The primary antibody (IPPF antibody) used was rabbit antiserum, whereas the secondary antibody utilized was alkaline phosphatase-labeled primary antibody.
2.7 Western blot analysis of IPPF expression in flaxseed protein
The contents of flaxseed proteins in all samples were determined and diluted to 1 µg/mL. Then, 5 mL of the diluted samples was mixed with an equal volume of the sample buffer (20% glycerol, 4% sodium dodecyl sulfate [SDS], 3% dithiothreitol, 0.002% bromophenol blue, and 0.125 M Tris-HCl, pH 6.8) and incubated at 98°C for 5 min to denature the proteins. The thermally denatured samples (10 µL/lane, equal to 5 µg/lane) were applied to 10% SDS-PAGE gels for 1 h at 50 mA and transferred onto a PVDF membrane (Hybond-P, GE Healthcare) for 1.5 h at 15 mA. The membrane was then cut into two pieces according to the molecular weights of the flaxseed proteins. Both pieces of membrane were blocked with 5% (w/v) skimmed milk in phosphate-buffered saline with Tween® 20 (PBST) at room temperature for 1 h and then washed twice for 2 min, once for 15 min, and thrice for 5 min with PBST. The membrane with IPPF was probed with the primary antibody anti-IPPF (which was prepared in 2.3) and the secondary antibody at room temperature for 1 h. After the specimen was washed three times, once for 2 min, twice for 15 min, and thrice for 5 min with PBST, the proteins on the membranes were detected with ECL prime detection reagents (GE Healthcare, IL, US) and Image Quant LAS 4000 (GE Healthcare). Densitometry was performed to determine the amount of phosphorylation by using Image Quant TL 7.0 (GE Healthcare).
2.8 Caco-2 and HepG2 cells culture
Caco-2 and HepG2 cells were removed from liquid nitrogen and quickly placed in a preexisting tube after complete dissolution. The cells were obtained under sterile conditions and inoculated to 8–10 mL containing 90% MEM + 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 µg/mL). The culture medium was cultured in a CO2 incubator at 37°C. After 24 h, the culture medium was replaced, and the growth state of the cells was observed.
2.9 Measurement of cell viability
Caco-2 and HepG2 cells were digested by trypsin when the confluence reached 80%. The cells were counted, and the cell concentration was adjusted to 2×104 cells/mL. The cells were seeded into a 12-well Transwell insert culture plate with 100 µL of cells per well. Different concentrations of 100 µL of IPPF were added per well and cultivated for 48 h. After incubation, the cells were washed twice with PBS; then, 10 µL of CCK-8 solution was added to each well and incubated at 37°C for 1–4 h. OD at 450 nm was measured, and cell viability was calculated. The appropriate concentration was screened. The experiment had four groups: control; low-IPPF-dose group (Low, 200 µg/mL IPPF); medium-IPPF-dose group (Medium, 400 µg/mL IPPF); and high-IPPF-dose group (High, 800 µg/mL IPPF).
2.10 Establishment of a Caco-2 cell monolayer model in vitro [13]
Caco-2 cells were digested with trypsin when the confluence reached 80%. The cells were counted, and cell concentration was adjusted to 8×104 cells/mL. The cells were seeded into a 12-well Transwell insert culture plate with 500 µL of cells per well, and 1 mL of the culture solution was added to the basal side. The culture media were changed 24 h after inoculation, and the culture medium was changed every 3 days. Transepithelial electrical resistance (TEER) was measured using Millicell ERS, and alkaline phosphatase (AKP) was determined using an AKP kit (NJJC, Nanjing, China).
2.11 Cholesterol transport contents of IPPF in Caco-2 cells
IPPF concentration was determined according to cell viability. The configuration of cholesterol micromicelles was determined in accordance with the slightly modified method of Zhang et al. [12]. In the apical side (AP)–basolateral side (BL) transport experiment, after the transport model was established, 0.4 mL of IPPF and cholesterol micelle solution was added to the AP side, and 1.2 mL of HBSS was added to the BL side. The 12-well Transwell TM culture plate was covered and then shaken at 50 r/min at 37°C. Then, 0.1 mL of the receiving solution was collected, and 0.1 mL of the blank receiving solution was added. Subsequently, 0.5 mL of HBSS was added to wash the cell layer three times. The cells were lysed with lysate on ice. Afterward, the lysate was sonicated to make a uniform solution. A 22-NBD-cholesterol kit (NJJC, Nanjing, China) was used to determine the contents of cholesterol-transport contents.
2.12 Western blot analysis of Caco-2 cells
The protein levels of NPC1L1 and ABCG5/8 in the microsomal fraction were determined through Western blot analysis with β-actin as an internal reference. Cells were lysed by cell lysate to extract proteins. The specific experimental method was similar to that described Sect. 2.7.
2.13 Determination of HepG2 cellular total cholesterol contents
First, HepG2 cells in a 24-well plate at a density of 2.5×105 cells/well were treated with different IPPF concentrations for 24 h. After each treatment, the total cholesterol of different groups was determined by a cholesterol kit (NJJC, Nanjing, China) following the manufacturer’s protocol.
2.14 Determination of mRNA levels in HepG2 cells
Total RNA was isolated from HepG2 cells by using the RNeasy mini kit (NJJC, Nanjing, China) following the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 1.0 µg total RNA by using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Berlin, Germany). Gene expression levels were analyzed via quantitative real-time reverse transcription polymerase chain reaction with a SYBR® Premix EX Taq II Kit and a Thermal Cycler Mx3000P QPCR System. mRNA levels were normalized using the β-actin (Actb) gene as an internal standard. The primer sequences and annealing temperatures used for the analysis are shown in Table 1.
Table 1
Primers for real-time RT-PCR
| Gene name | Forward primer | Reverse primer |
| Actb | TCAGGTCATCACTATCGGCA | TCATGGATGCCACAGGATTC |
| Hmgcr | TGTGGGAACGGTGACACTTA | GCTGACGCAGGTTCTGGAA |
| SREBP-2 | CTGGCAAATCAAAAGAACAAGC | GGACATTCTGATTAAAGTCCTCGA |
| LXR-a | ATGGACACCTACATGCGTC | CTTCAGGCGGATCTGTTCTT |
| CYP7A1 | CTGTCATACCACAAAGTCTTATGTCA | ATGCTTCTGTGTCCAAATGCC |
| Soat1 | GATGGGGTTATGTTGCTATGC | GGGCTCCTGTTTGATATTCCG |
2.15 Statistical analysis
The experiments were conducted in triplicate, and data were expressed as mean ± standard deviation. One-way ANOVA and Duncan’s new multiple-range test were performed to determine significant differences among the means at p < 0.05.
{kind=link}