Cell lines HSC-3 and HSC-4 were purchased from the Japanese Collection of Research Bioresources (JCRB, Shinjuku, Japan) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Sigma-Aldrich, St. Louis, MO) with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and penicillin and streptomycin (Thermo Fisher Scientific) at 37°C with 5% CO2.
Wound healing assay, cell migration, and invasion assays
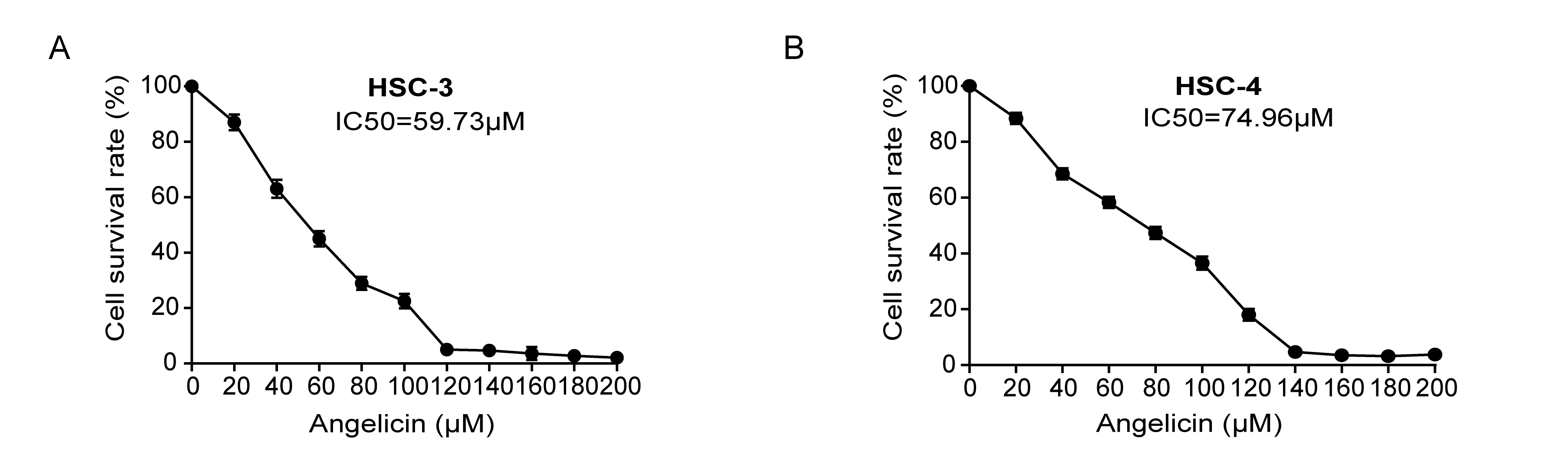
To measure cell mobility, we used a wound healing assay: 4×105 cells per well seeded in 6-well plates were incubated for 18–24 h and scratched with a 200 µl pipette tip. After using phosphate buffer saline to wash the wells thrice, wound healing photographs were taken at the same position as the scratching area at 0 h and 12 h after scratching.
To determine cell migration and invasion, we used a transwell system, including 8-µm pore filters (Corning, New York, NY, USA) and 24 well plates. For invasion assays, the bottom of the upper transwell chamber was coated with Matrigel (BD Bioscience, San Diego, CA, USA) for 1 h at 37°C. The upper chamber was seeded with 1×105 cells in a 200 µl serum-free basic DMEM medium. The lower chamber was filled with 500 µl complete culture medium and angelicin and the concentration used was IC50. After incubating for 24 h at 37°C, the cells on the lower surface of the transwell membrane were fixed by methanol and stained with 0.5% crystal violet. Five randomly selected fields were photographed and counted per well.
Cell cycle and apoptosis analysis
Cells were treated with angelicin for 24 h and collected. For cell cycle analysis, cells were fixed by cool 70% ethanol at 4°C overnight and stained with propidium iodide (PI) and RNase (KeyGEN BioTECH, Nanjing, China). The percentage of cells in the G1, G2, and S phases was analyzed. For cell apoptosis analysis, cells were incubated with Annexin V and PI per the manufacturer’s instructions (4A Biotech Co., Ltd., Beijing, China). Next, the percentage of Annexin V+PI−and Annexin V+ PI+ cells were detected as the apoptosis rate. All samples were analyzed by flow cytometry (Milipore Guava, St. Louis, Missouri, USA).
Differentially expressed genes analysis
TRIzol was used to extract total RNA. A NanoDrop 2000 spectrophotometer (Thermo Scientific, USA) was used to evaluated RNA purity and quantification. An Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) was used to test RNA integrity. Next, the TruSeq Stranded mRNA LTSample Prep Kit (Illumina, San Diego, CA, USA) was applied to construct the total RAN-seq libraries. An Illumina HiSeq X Ten platform was used to sequence the libraries and generated150 bp paired-end reads. To obtain the clean reads, we used a Trimmomatic [22] to remove the low quality reads from raw reads. OE Biotech Co., Ltd. (Shanghai, China) conducted the transcriptome sequencing and analysis. The clean reads to the human genome (GRCh38) were mapped by HISAT2 (version 2.1.0) [23], the fragmentsper kilobase per Million (FPKM) of each gene was calculated by Cufflinks [24], and the read counts of each gene were obtained by HTSeq-count [25]. The DESeq2 R package was used to performed differential expression analysis [26]. The threshold for the significantly differential expression was p value < 0.05 and |log2foldchange (FC) | > 2.
RNA extraction and RT-qPCR
The total RNA of cells was isolated by using TRIzol (Invitrogen, Carlsbad, CA, USA). The RT reagent Kit gDNA Eraser (TaKaRa Bio, Kusatsu, Shiga) was applied to prepare cDNA samples. The
abundance of transcripts was detected by SYBR-Green (TaKaRa Bio) and used GAPDH as an internal reference. The primers observed were DUSP6 : F: 5༇-GAGTCTGACCTTGACCGAGACCCC − 3′, R:5′- TTCCTCCAACACGTCCAAGTTGGT-3′;c-MYC: F: 5༇-GCCAAGCTCGTCTCAGAGAAG − 3′, R:5′-CAGAAGGTGATCCAGACTCTG − 3′; GAPDH: F: 5༇-ACAACTTTGGTATCGTGGAAGG-3༇, and R:5༇- GCCATCACGCCACAGTTTC-3༇.
Western blot
Total proteins were obtained by RIPA lysis buffer (Solarbio, Beijing, China) containing PMSF (Solarbio). 10% tris-polyacrylamide gels (SDS–PAGE, Bio-Rad, USA) were loaded with 20 µg of cell protein lysate, which were transferred to 0.45 µM PVDF membranes (Solarbio). After incubation with primary antibodies at 4°C overnight, the membrane was incubated with secondary antibodies (ZSGB-BIO, Beijing, China) at room temperature for 2 h. Primary antibodies were used as the following dilutions: Anti-DUSP6 (Abcam, Cambridge, MA, USA), 1:1000; Anti-c-MYC (Abcam), 1:1000; GAPDH (Abcam), 1:3000.
Immunohistochemistry (IHC)
Human tissues and xenograft tumor were fixed in 4% polyformaldehyde followed by paraffin embedding. IHC staining was executed according to the standard protocol of the manufacturers. In summary, the epitope retrieval of DUSP6 was performed with Tris-EDTA buffer (pH 9), and the one of c-MYC was 10 mM sodium citrate (pH 6). Normal sheep serum (ZLI-9056), (ZSGB-BIO) was used to block non-specific antibody binding. During the staining, a rabbit reinforced polymer detection system (PV-9001), (ZSGB-BIO) and GTVision TM +detection system (GK600505; Gene Tech, Shanghai, China) were used. The primary antibodies used were anti-DUSP6 (ab76310, Abcam) and anti-c-MYC, (ab32072, Abcam). All results were semi-quantified based on average optical density (AOD).
Biolayer Interferometry (BLI)
BLI assays and data processing were used (Octet Red 96 system [ForteBio, USA]). First, recombinant human DUSP6 protein with his tag N-Termius (ab183239, Abcam) was diluted to 100 µg/ml by using TBS buffer. The running buffer was DMSO (10% in TBS). The small molecular drug angelicin was diluted into four concentrations—12.5 µM, 25 µM, 50 µM, and 100 µM—in running buffer. The Ni-NTA sensor was used, and the time of association and dissociation was 60 s and 120 s, respectively. Last, the binding affinity was detected by the affinity dissociation constant (KD).
Mouse xenograft model
Female BALB/c nude mice received subcutaneous injections of HSC-3 cells (1 × 107) in the right armpit at the age of 6 weeks. Mice were divided into two groups (n = 6 mice/group). The angelicin group was treated with angelicin, dissolved in corn oil (Yuanye Bio-Technology, Shanghai, China), at a concentration of 20 mg/kg per day delivered by intraperitoneal injection; the control group was treated with an equal volume of corn oil. The mice were treated with angelicin for 20 consecutive days from the fifth day after injection of HSC-3 cells; substantially, the mice were sacrificed. Normal epithelial tissue of mice was used as the IHC normal group. Tumor volumes were measured every 2 days. The formula V (mm3) = 0.5 × length (mm) × width2 (mm2) was used to calculate tumor volume.
BALB/c nude mice were purchased from the Vital River company (Beijing, China). The study was approved by the Ethics Committee of West China Hospital of Stomatology, Sichuan University.
{kind=link}
{kind=link}