Fusarium wilt is a vital ailment of pigeonpea and is realized to be the worst problem for cultivation of this crop in India [28]. The annual economic losses due to wilt in pigeonpea have been assessed at US $ 36 million in India and the subsequent yield loss can also be increased up to 100% in susceptible cultivars [29]. So, it is necessary to find a way out in sustainable manner to prevent the disease in this crop. Among the different control measures, early diagnosis and control in the light of advancement of molecular technologies are quite promising. The importance of wide level of survey is not only related to epidemiology of a disease to understand, describe, compare and predict epidemic, but also make a chance to explore the most promising highly tolerant genotype/s against a particular disease from surveyed areas. From 1980 to till date, a number of surveys were carried out in different pigeonpea growing countries. The present survey-based study reported regional diversity of more than 25 pigeonpea-cultivated areas representing the major agro-climatic regions of India under pigeonpea production, which were found to be suitable for growing local genotypes/cultivars due to their potential nature of tolerance/susceptibility mechanism to FW. Taking into consideration of the most variable geographical region, MP represented a great diversification in disease incidence range from resistant to highly susceptible areas. As far as lowest disease epidemiology is concerned, TN was found to be the lowest disease prone state with no class as highly infested area and very highly infested area. Besides India, this disease was reported from East Africa and Malawi, where yield losses were reported to have crossed above 50% in some places. Yield losses in pigeonpea due to FW was also reported form countries like Bangladesh, Indonesia, Grenada, Myanmar, Mauritius, Nevis, Nepal, Venezuela, Trinidad and Tobago [30] This pathogen was also recorded for causing severe disease in the Southern Zambezia province under South Africa [31].
The study of exploration of pigeonpea genotypes to FW displayed an overall contrasting response for all selected genotypes. The experimental reference variety ICP–8863 displayed a negligible or near to zero level of symptoms (D.I–3.33%) in one out of 30 plants as it is already reported for its best resistance mechanism against FW independent to environmental and geographical conditions. It is finally concluded that Richa was one of the most highly tolerant genotypes (%D. I- 6.67) after ICP–8863 with a higher degree of tolerance potential and that could be due to specific morphological modification in the xylem vessel of the root system or targeted activation of some defense related proteins and transcription factors during fungal stress or due to both reasons. On other hand, Parwati was authenticated as one of the most susceptible among all experimental genotypes and it could be due to its weak defense system. Although ICP 8863 and ICP2376 were confirmed for their extreme resistance and susceptible characteristics, respectively in our experiment and were earlier validated so many times for these features [28, 32]. Therefore, these three genotypes viz. ICP 8863, ICP2376 and Richa were picked for further molecular biology studies. In overall evaluation data, all the four classes of disease scales were fitted for selected genotype population. A number of efforts have been taken to find out the most potential genotypes or cultivars resistant against FW over time and pathogenic strains. First time observations on wilt resistance in pigeonpea was reported by Butler in 1908 [33]. Apart from this, 950 genotypes screened had shown zero resistance to FW with less than 10% wilt incidence in 19 genotypes [29], 16 out of 31 pigeonpea cultivars screened were resistant to wilt with highest degree of resistance (2.15 %) in BWR 369 cultivar [34]; six cultivars were reported to be resistant to FW among 216 late maturing pigeonpea germplasm evaluated [7]; resistance screening in 976 genotypes, germplasm and breeding lines FW using wilt sick plot confirmed 92 genotypes resistant after a rigorous two years testing at Patancheru, India [35]. Another study on evaluation of new elite pigeonpea germplasm using wilt sick plots demonstrated a consistent rate of high level of resistance (DI < 20.0 %) in ICEAP–00040 genotype to FW reported in Kenya, Malawi and Tanzania [31].
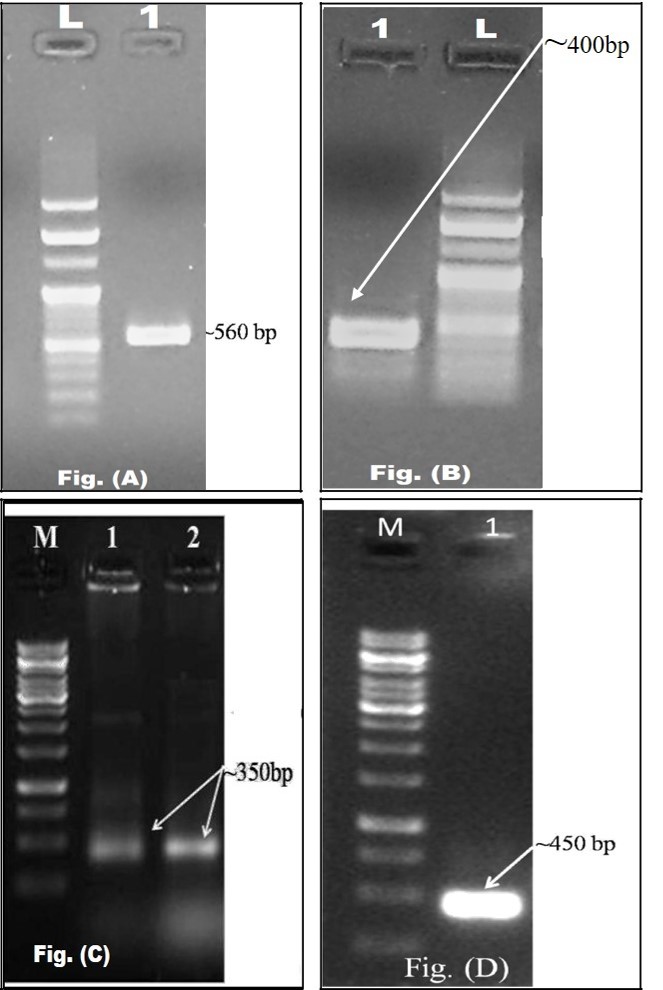
For the identification, quantification and expression profiling of highly active plant defense responsive genes, proteins under enzyme group and in most cases, transcription factors and high throughput molecular techniques have been developed and customized day by day for their successful implication. In our current study, understanding of spatio-temporal expression of major defense responsive promising genes during pathogenesis and the study of different patterns of symptomatology based tolerance in various plant genotypes specific to various agro-climatic regions of India can provide the vital knowledge for crop improvement with well-developed tolerance mechanism. Higher plants directly defend a number of biotic and abiotic stresses with their broad range defensive mechanism, especially the production and accumulation of pathogenesis related proteins in plants in response to invading pathogen [36]. A wheat β–1,3-glucanase gene (TaGlu) induced by Puccinia striiformis (Pst) was cloned, characterized and qPCR analyses confirmed that the transcription of TaGlu was induced in both compatible and incompatible interactions [13]. In our present molecular biology study, the modified RNA extraction method is equally useful to extract stressed sample as well as fungal RNA with maximum reproducibility to trace their gene’s functions and behaviour under various stresses imposed during pathogen attack [16]. By using isolated RNA from modified method, RT-PCR based detection of four genes belonging to AO enzyme and PR protein groups was performed and their successful amplifications made an inference that the genes are actively present in plant system during FW stress. The corresponding mRNAs were synthesized at the nucleus and circulated over cytoplasm followed by their translation to the consequent proteins or enzymes, which are directly or indirectly involved in defense response to significantly enhance their tolerance to pathogen attack. To precisely monitor the expression level of a gene at various stages, qPCR-based transcript profiling approach is highly adorable and the present comparative analysis of expression of AO genes with PR protein genes has revealed a variable mode of expression between susceptible and biologically validated resistant cultivars. It was also indicated that no significant differential induction has been located in case of SOD for both genotypes and CHS for ICP2376. It can be concluded that these genes did not have any significant expression variation under FW stress by considering the varietal difference. Higher magnitude of transcript expression at the last TP confirms that the expression was specific to infection stage rather than gene and genotype. This may help the plant to cope with the deleterious effects of pathogen attack at early stage critical for later growth and development [37]. Comparatively, the imperative variations were observed in the case of APX and β–1, 3 glucanase genes. An attractive finding of the present study is the differential expression of CHS gene activity in susceptible and resistant (biologically validated) pigeonpea genotypes whereas the induction level of β–1,3-glucanase activity was found increasing in Richa genotype under continuous pathogen load. Liu et al. [13] and Nagy et al. [38] also reported a similar type of expression behaviour of β–1, 3-glucanase gene induced by stripe rust pathogen Puccinia striiformis f. sp. Tritici in wheat and CHS gene with an induced response to pathogen infection in Norway spruce phloem respectively. It is known that plants possess inducible and constitutive defences. Structural analysis of PR-genes and antioxidant genes suggests that they are active in signalling cascade(s) that coordinate initial plant defense responses in order to impair pathogen growth. Besides, PR-gene products may have a function in plant development and therefore, be expressed in challenging but tolerant genotypes of pigeonpea plants, ready to detect any attack. Our results suggest that both mechanisms may be important for FU tolerance. The difference between tolerance and susceptibility depends on a number of internal and external factors of which early detection of pathogen is considered as prime factor.
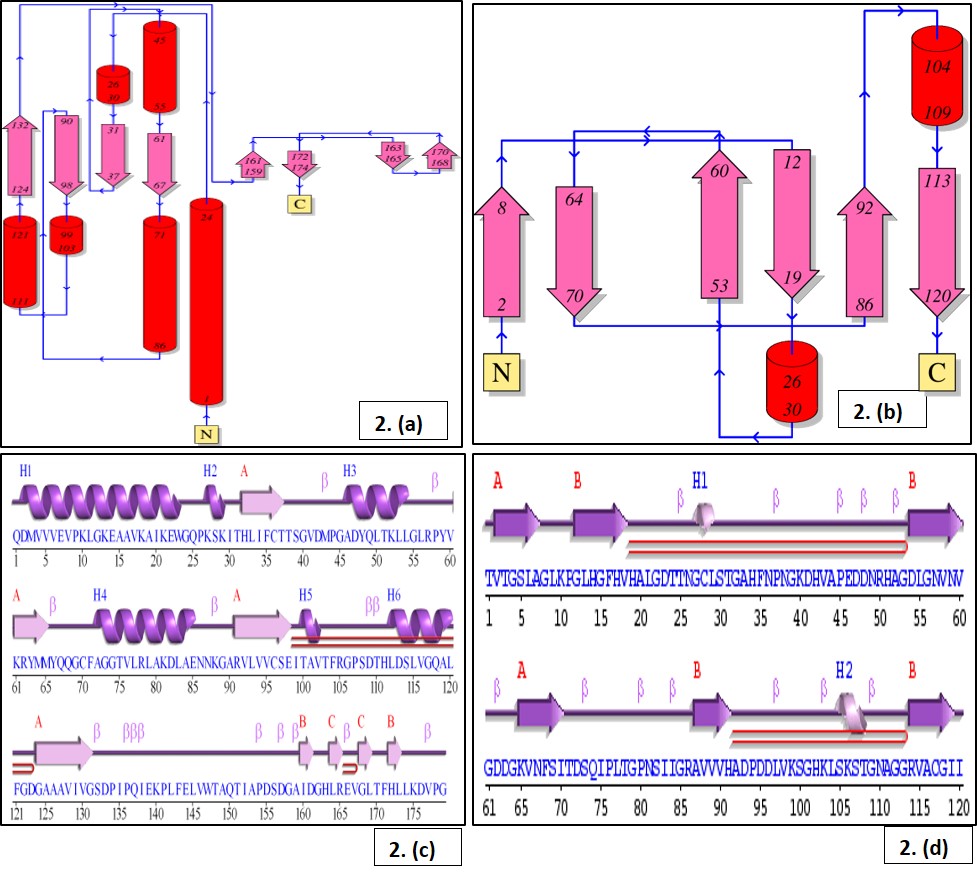
The in-silico 3D structure was predicted for the proteins (CHS and SOD). The value of instability index (CHS: 28.02 and SOD: 5.66) indicated their high stability as a model. GMQE score value of CHS (0.99) and SOD (0.93) indicated the expected accuracy of the model from target template alignment and target sequence coverage. Higher GMQE value indicated higher reliability. QMEAN value provides estimate of ‘degree of nativeness’ of protein structural features in the model on a global scale. It ranges from –4 to 0. Assessed QMEAN values, 0.84 and 0.85 for CHS and SOD respectively were within the acceptable range of high-quality model. Further validation and refinements of model were confirmed using Ramachandran plot. The phi (φ)/psi(ψ) angles of the amino acid residues revealed that both CHS and SOD proteins hold more than 90% amino acids in favoured region, thus considered accurate. Identified active site in CHS (Cys164), active-site nucleophile is reported to be crucial in polyketide formation and initiation series of decarboxylation, condensation, and cyclization reactions along with other amino acids [39,40]. SOD shield cells from ROS by catalysing the disproportionation of superoxide anion radicals into molecular oxygen and hydrogen peroxide. Predicted active site Copper/Zinc superoxide dismutase signature 1 and 2 at regions 15–25 and 109–120 contributes to stability of the framework and dimer assembly [41]. The maximum correlation between docking score were considered strongest protein-ligand interaction. Thus, both the CHS and SOD proteins bind with CBH-c actively in order to block the functional activity.
{kind=link}
{kind=link}