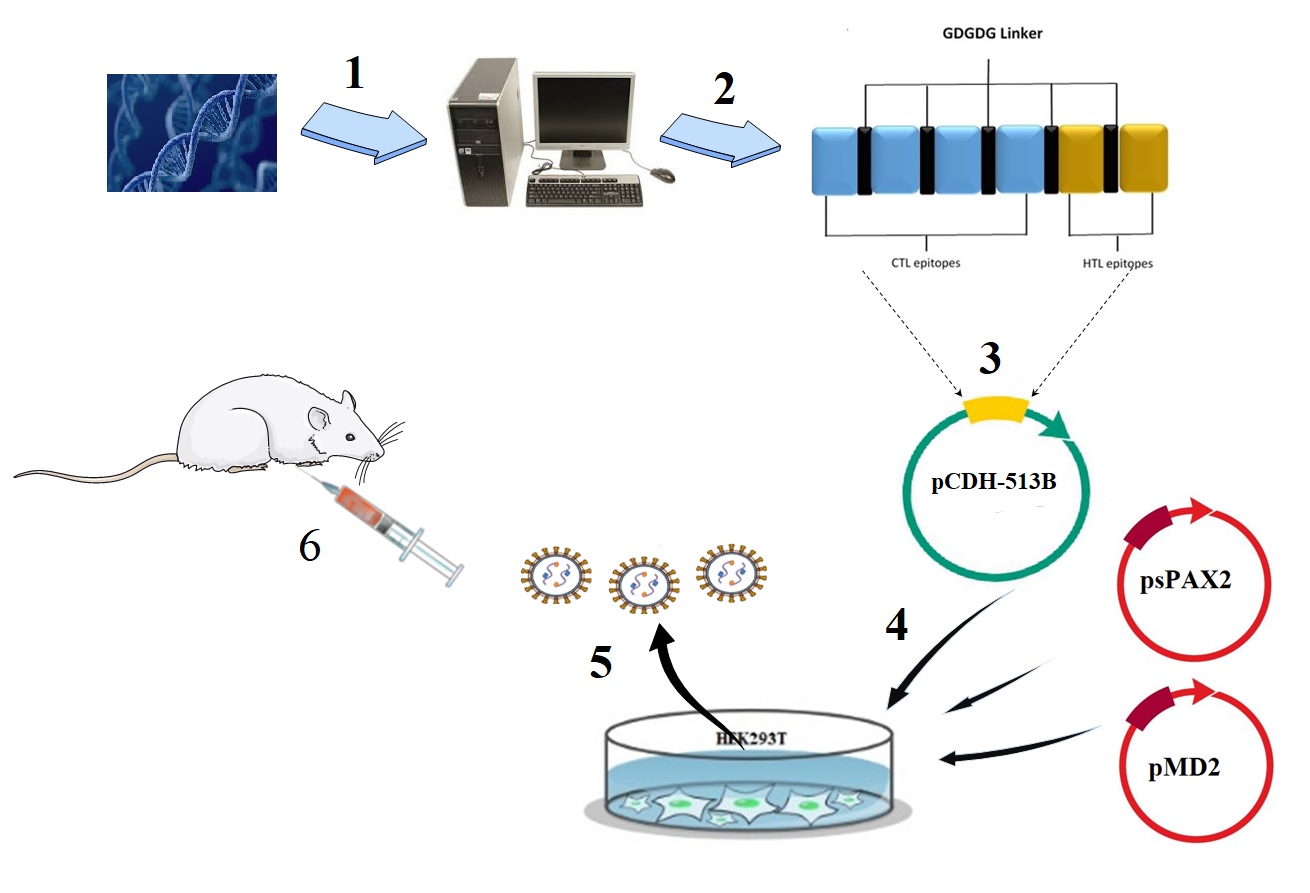
Design of multi-epitope lentiviral vaccine
The protein sequences of KMP11 and HASPB of L.major (Friedlin strain) were obtained from GenBank, protein database of the National Center for biotechnology information (NCBI, http://www.ncbi.nih.gov/genbank/). The ID of KMP11 and HASPB was AAR84616.1 and CAB39972.1, respectively. Then, with the help of immunoinformatics software such as T- cell epitope prediction, B-cell epitope prediction, docking studies and molecular dynamics simulation, the epitopes of KMP11 and HASPB proteins were selected in silico space (15). Also, using GDGDG linker, final multi-epitope vaccine was designed (Figure. 1).
Cell culture
HEK293T cell line (Human Embryonic Kidney, ATCC CRL-3216) was purchased from Mede Bioeconomy Company Iran and seeded in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Darmstadt, Germany). The cells were supplemented with 10% foetal bovine serum (FBS) (Gibco, USA), penicillin (100 units/ml)/streptomycin (100 mg/ml) (Invitrogen, Carlsbad, CA, USA), finally the cells were incubated in a humidified atmosphere with 5% CO2 at 37°C.
Synthesis and confirmation of the pCDH-multi-epitope construct
The designed multi-epitope was synthesized by Genscript, USA, and merged into pUC57 plasmid. Then, the construct was subcloned into pCDH513B lentiviral vector using XbaI and BamHI restriction enzymes. The recombinant pCDH-multi-epitope was digested using XbaI and ApaI and sequenced. In this construct, to express multi-epitope cytomegalovirus (CMV) promoter was used. Also, GFP and puromycine were expressed using eukaryotic elongation factor (eEF) promoter. Moreover, GFP and puromycine were separated by T2A factor. As a result, transfer vector expressed multi-epitope, GFP and puromycine resistance factor from two mRNAs (Figure. 2).
The pCDH-multi-epitope plasmid was used to transform the competent E. coli DH5α cells. After transformation of bacteria, the LB agar containing ampicillin (100 µg/ml) was used to bacteria culture at 37°C for 24 h. Then, from the colonies appeared on the plate, plasmid extraction was carried out using Miniprep plasmid extraction kit (Qiagen, Germany) according to manufacturer’s protocol. Afterward, the restriction enzymes containing XbaI and ApaI were used to digest the extracted plasmid and electrophoresed on 1% agarose gel. Finally, sequencing of plasmid was done to further confirm the cloning. Also, in order to extract of PAX, MD, and pCDH lentiviral plasmids, all procedures were performed.
Virus production (rLV-multi-epitope and rLV-empty)
By using the transfer lentiviral vector (pCDH-multi-epitope or pCDH-empty) and two PAX and MD plasmids, transfection of HEK293T cells were done based on Trono protocol (calcium phosphate transfection method)(16). On the first day, 3×106 HEK293T cells were seeded in a 10-cm plate in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA), supplemented with 10% FBS (Gibco, USA). On the second day, a mixture of 21 μg of the transfer vector (pCDH-multi-epitope or pCDH-empty), 21 μg of psPAX2 vector and 10 μg of pMD2.G vector with transfection buffer was added dropwise to the cells. Then, 14 hours’ post-transfection, fresh medium was used to replace the previous medium. After 24 hours, with the help of a fluorescent microscope, evaluation of GFP expression was performed. Moreover, 24, 48 and 72 hours’ post-transfection the supernatant of the packaged lentivirus (named thereafter rLV-multi-epitope or rLV- empty) were harvested and centrifugation at 350 g at 4°C for 5 minute was done. Moreover, the supernatant was filtered through a 0.22-μm filter. Finally, the rLV-multi-epitope or rLV-empty were stored at −70°C for next experiments.
Titarion of virus
Using flow cytometry, the number of GFP-positive cells were counted and, the titre of packaged viruses was defined. See the virus count formula
TU/mL= (1∕4) (F ∗N∗D∗1000) ∕V, Where F = percentage of fluorescent cells (EGFP), N = number of cells at the time of transduction (about 1×105 cells per well), D = fold dilution of vector sample used for transduction and V = volume (mL) of diluted vector sample added into each well for transduction. All steps have been previously described(17).
Western blot
In order to confirm the expression of rLV-multi-epitope, western blot was carried out (18).
The First, the rLV-multi-epitope and rLV-empty were used to transduce of HEK293T cells. Thereafter, the cells were treated with 2 μg/mL puromycin (Sigma-Aldrich, USA). Subsequently, the transduced HEK293T cells (rHEK-multi-epitope, rHEK-empy) and non-transduced HEK293T cells (HEK-non) were seeded in 6-well plates. Then, after 72 h, the cells were trypsinized and centrifuged at 190g for 5 min at 4 °C. Also, the cells were lysed in 200μL of protease inhibitor-containing RIPA buffer (Thermo Fisher Scientific, IL). The lysates were centrifuged at 8800g for 12 min at 4 °C. Then, the protein concentration of the supernatants was determined using a BCA Protein Assay Kit (Thermo Fisher Scientific, IL). Lysates/lane of 30 μg were loaded onto a 12% SDS–PAGE gel and then transferred onto a nitrocellulose membrane (Bio-Rad Laboratories, CA).
Thereafter, the protein was transferred onto a nitrocellulose membrane (Bio-Rad Laboratories, CA). Then, using 2.5% non-fat milk, the membranes were blocked and using Anti-polyHistidine antibody (anti-His antibody) (Abcam, USA) conjugated with horseradish peroxidase (HRP) at 1:2000 dilution immunoblotting was performed.
Finally, to visualize the protein band, the substrate of the tetramethylbenzidine (TMB) was used. Finally, the optical density (IOD) of the band was obtained and it was integrated.
Immunization of BALB/c mice
Experiments were performed on twenty-four female inbred BALB/c mice, 6-week-old which were purchased from Pasteur Institute of Iran and were maintained in the Institutional animal care of the Fasa University of medical sciences based on the specific National Ethical Guidelines for Biomedical Research issued by the Research and Technology Deputy of Ministry of Health and Medicinal Education (MOHME) of Iran (2005).
Three groups of mice were considered and each group was randomly received 8 mice (n= 8), group 1 (rLV-multi-epitope), group 2 (rLV-empty) and group 3 (PBS). The injections were carried out subcutaneously twice during the 1 weeks (with a 3-day interval) with 1×106 TU of rLV-multi-epitope (3×106TU/mL) to group 1, and 1×106 TU of rLV-empty (1×107 TU/ mL) and 0.1 mL PBS to control groups. Finally, three weeks post-immunization of mice, cytokine assay was done.
Parasite culture
In order to obtain a sufficient number of L. major (MHRO/IR/75/ER (parasite, promastigote parasites were cultured and enriched in RPMI 1640 (Gibco, UK) supplemented with 20% foetal calf serum (Gibco, Darmstadt, Germany), 100IU/ml of penicillin and 100 μg/mL of streptomycin. 100 IU/mL of penicillin and 100 μg/mL of streptomycin to obtain an adequate number of promastigotes, as described by Dehkordi et al (19).
Antibody analysis
To investigate the humoral immune response, three-week post-injection the sera of vaccinated mice were collected and analyzed in triplicate as previously described. To determine the presence of antibodies, 96-well flat-bottom plates were coated with L. major promastigotes that there was obtained from stationary phase of culture. To block the plates, 1% (w/v) BSA was used. Also, mice sera were diluted 1:100 and applied to the plate. Then, detection of attached antibodies was done, and the plate was incubated with anti-His antibody (Abcam, USA) and developed with peroxidase substrate tetramethylbenzidine (TMB). Finally, we used an ELISA microplate reader (Bio-Rad) to read the plate at 410 nm.
Cytokine analysis
In order to evaluate the level of cytokine expression, the splenocytes of mice were separated and analyzed as previously complained. Three weeks after injection, in aseptic conditions the spleens of immunized mice and control groups were separated and homogenized. Then, the splenocytes were recovered, washed three times, and seeded in triplicate in a 96-well plate. Moreover, the cells were stimulated using L. major lysate, Soluble Leishmania Antigen (SLA).
Finally, to determine the levels of IL-4 and IFN-γ, after 72 hours Supernatants were harvested and mentioned cytokines were assayed in triplicate by using sandwich ELISA kits (Abcam, USA) based on the manufacturer’s protocol.
Statistical analysis
According to Mann-Whitney U-test, data analysis was performed and SPSS (V.18). A P-value < 0.05 was considered as statistically significant.
{kind=link}