Materials
Hyaluronic acid (HA, 97%) and glycidyl methacrylate (GMA, 98%) were purchased from Maclin Biochemical Co. Ltd and J&K Chemical Ltd, respectively. 2-hydroxy-4’-(2-hydroxyethyl)-2-methylpropiophenone (Photoinitiator Irgacure 2959, 99%) was obtained from Aladdin Bio-Chem Technology Co. Ltd. All chemicals were used as received without further purification.
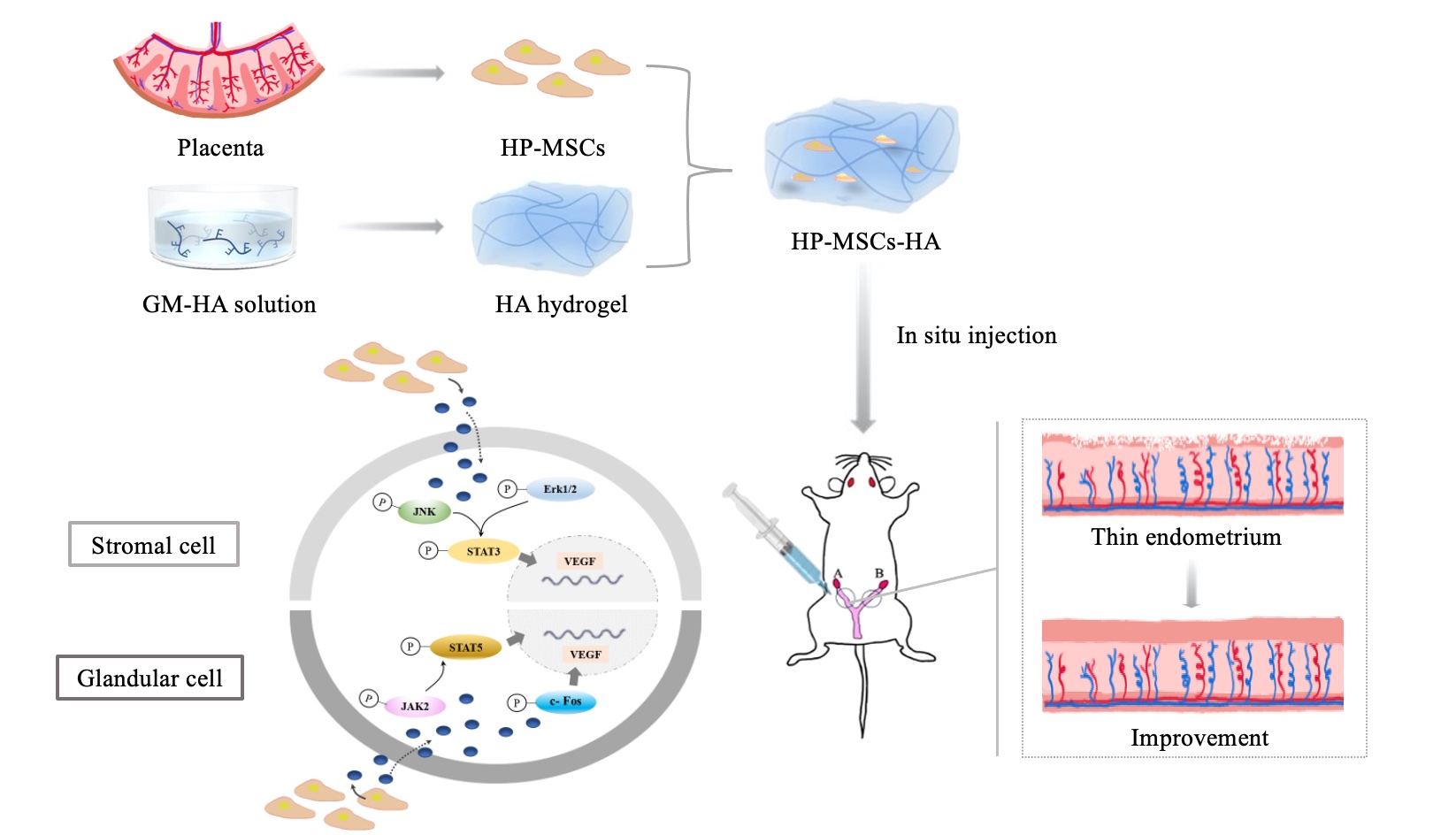
Identified HP-MSCs were kindly provided by the College of Life Sciences-iCell Biotechnology Regenerative Biomedicine Laboratory, Zhejiang University. The identification results of these HP-MSCs were provided in supplement materials. The cells were cultured in basic DMEM/F12 medium (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS, Biological Industries, Kibbutz Beit-Haemek, Israel), 1% penicillin (Geno, Hangzhou, China) at 5% CO2 and 37 oC as previously optimized [29, 30].
Preparation and characterization of HA hydrogel
The polymerizable HA was prepared via the chemical modification of HA by using GMA, as shown in Figure 1A. Briefly, 0.5 g HA was dissolved in 100 mL mixture of N, N-dimethylformamide (DMF), and phosphate buffer saline (PBS) solution (0.01 M, pH = 7.40) with a volume ratio of 1:1 overnight. The mixture was subsequently mixed with 3.35 g of triethylamine (TEA) for 30 min. Then, 6.65 g GMA was added to the mixture and stirred for 5 days at room temperature. The reaction mixture was precipitated in 20 times excess acetone solution. The precipitates were collected and dried in a vacuum oven. The products were purified by dialysis in deionized water for 3 days and further dried by lyophilization, leading to the final GMA functionalized HA powders, coded as GM-HA. To prepare HA hydrogel, 10 mg GM-HA were dissolved in 1 mL aqueous solution containing 1‰ Irgacure 2959 photoinitiator. The mixed solution was then exposed to 365 nm UV light for 15 min, leading to the formation of HA hydrogels.
1H-NMR spectrum of GM-HA was recorded on a 400 MHz Bruker NMR instrument with D2O as a solvent. Fourier transform infrared (FT-IR) spectra of GM-HA and HA hydrogel were obtained by a Bruker Vector 22 spectrometer. The rheological measurement of HA hydrogel was carried out on DHR2-2183 rheometer at 37 oC. The morphologies of HA and HA hydrogel were observed by Nova Nano scanning electron microscopy (SEM 450, Thermo FEI). The HA and lyophilized HA hydrogel power were evenly spread on the conductive glue and purged three times with an ear wash ball for SEM samples. The prepared samples were sprayed with platinum for 60 s before SEM observation. After that, 500 µL HA hydrogel was mixed with 4 ´106 HP-MSCs and incubated at 37 °C for 10 min to prepare HP-MSCs-HA, and the mouse was injected with 25 µL HP-MSCs-HA per uterus.
Isolation and culture of Human stromal and glandular cells
A total of 20 female patients aged between 24-48 years old with normal menstrual cycles (21-35 days) were recruited at Women’s Hospital of Zhejiang University, China. They haven’t received hormone therapy at least three months before surgery. All the patients underwent hysteroscopy and endometrial biopsy during the infertility examination. Stromal and glandular cells were isolated from the endometrium as previously described [31]. Both groups of cells were cultured in DMEM/F12 medium containing 10% FBS and 1% Penicillin-Streptomycin at 37 oC in a humidified environment with 5% CO2. After coculturing with HP-MSCs for 24 h, 48 h, 72 h, respectively, the cells were used to measure EdU/Transwell/Western blot assay.
In vivo tracing of CM-DiD/CM-DiR-labeled HP-MSCs
The commercial cell membrane red fluorescent probe CM-DiD/CM-DiR (US Everbright® Inc, Jiangsu, China) was diluted with DMSO according to the manufacturer’s instructions to make a final concentration of 1 mM. HP-MSCs at a density of 106 cells/mL were suspended in 5 mL PBS. 10 µL CM-DiD or CM-DiR cell-labeling solution was then added at 37 °C for post-instillation tracking in utero. After 20 min, the labeled cells were spun at 800 x g for 3 min and followed with washing twice with 5 mL PBS at 1000 x g for 5 min each. The CM-DiD and CM-DiR labeled HP-MSCs were then mixed with as-prepared HA hydrogel, respectively, to form two HP-MSCs-HA with different fluorescent labels. The mouse model was divided into two groups, as shown below, one group was injected with CM-DiR labeled HP-MSCs and HP-MSCs-HA for imaging, using an IVIS Spectrum to detect after transplantation for 1, 3, 7, 14, and 35 days, respectively. The other group was injected with CM-DiD label HP-MSCs and HP-MSCs-HA, which was used for the frozen section. Slides with 6 µm thick (cutting with a CryoStar NX50, Thermo) were observed by an Olympus IX81-FV1000 fluorescence microscope.
CM-DiR labeled group:
Left uteri: instillation of HP-MSCs alone (coded as HP-MSCs)
Right uteri: instillation of HP-MSCs-HA (coded as HP-MSCs-HA)
CM-DiD labeled group:
Left uteri: instillation of HP-MSCs alone (coded as HP-MSCs)
Right uteri: instillation of HP-MSCs-HA (coded as HP-MSCs-HA)
Transwell migration assay
Human stromal cells or glandular cells were cultured in the upper plate compartment (8-μm, 24-well insert, Corning, NY, USA), while the HP-MSCs were cultured in the lower chamber. They were then cocultured at 37 oC for 24 h, 48 h, and 72 h, respectively. The medium and cells were then removed from the upper chamber using cotton swabs with 1×PBS, while the migrated cells on the bottom surface of the membrane were fixed and stained with 0.5% crystal violet [32]. Cells from 5 random fields were counted using an inverted microscope with a magnification of 200x.
EdU proliferation assay
Human stromal cells or glandular cells were plated in 24-well plates and cocultured with HP-MSCs which were in the upper insert (0.4-μm, 24-well insert, Corning, NY, USA). After incubated for 24 h, 48 h, and 72 h, respectively, the transwell insert loaded with HP-MSCs were removed, and cell proliferation activity was assessed using a commercially available EdU Assay Kit (RIBOBIO, Guangdong, China) according to the protocol provided. Cell proliferation was quantified by the incorporation of EdU into the newly synthesized DNA of replicating cells. The proliferated cells were dyed red, while the nuclei of all cells were dyed blue with DAPI. By counting EdU/DAPI ratio, cell proliferation ability can be assessed.
Protein isolation and western blot analysis
Proteins were extracted from the treated human endometrial stromal cells and glandular cells and then lysed by RIPA lysis buffer (Cell Signaling Technology, Boston, MA, USA) as previously described [24]. The concentration of protein was detected using a bicinchoninic acid protein assay kit (Thermo Scientific, Waltham, MA, USA). Proteins were denatured in a 5× SDS-PAGE loading buffer (CWBIO, Beijing, China). Then they were separated on sodium dodecyl sulfate-polyacrylamide gels and subsequently transferred onto nitrocellulose membranes. After being blocked with 5% BSA (Albumin from bovine serum) in PBS, the membranes were incubated with primary and secondary antibodies. The immunoblots were washed with PBST (PBS with 0.1% Tween-20). And then the membranes were incubated overnight with primary antibodies at 4oC. After washing, secondary antibodies were added and incubated at room temperature for 1 h in the dark. Finally, the membranes were probed with an Odyssey CLx (LI-COR, USA). The signal intensity was calculated with ImageStudio. Protein expression was normalized to β-Actin.
Rabbit monoclonal anti-p-JNK (9255), rabbit monoclonal anti-JNK (9252), rabbit monoclonal anti-p-Stat3 (9145), mouse monoclonal anti-Stat3 (9139), rabbit monoclonal anti-p-Erk1/2 (4370), rabbit monoclonal anti-Erk1/2 (4695), rabbit monoclonal anti-p-Jak2 (3771), rabbit monoclonal anti-Jak2 (3230), rabbit monoclonal anti-Stat5 (D3N2B), rabbit monoclonal anti-p-c-Fos (5348), rabbit monoclonal anti-c-Fos (2250), rabbit monoclonal anti-p-c-Jun (3270), and rabbit monoclonal anti-c-Jun (9165) were purchased from Cell Signaling Technology. Mouse monoclonal anti-β-Actin (sc-47778) was purchased from Santa Cruz. Rabbit monoclonal anti-VEGF (GTX102643) was purchased from Gene Tex. Goat anti-mouse fluorescent antibody (926-68020) and Goat anti-rabbit fluorescent antibody (926-32211) were purchased from LI-COR (USA).
Establishment of the endometrium-injured mouse model
8-week-old female ICR mice of clean grade (SLAC company, Shanghai) were reared in the animal center under controlled conditions (21-24 oC, relative humidity 40-60%, 12 hours light / 12 hours dark cycle), with free access to food and water. To mimic endometrial injury in the clinical setting, we established a mouse model of thin endometrium by both mechanical and chemical injury at the estrous period. Briefly, the surgery involves mechanical intrauterine operation with a syringe contacting with the uterine cavity, as well as chemical injury by intrauterine perfusion with ethanol(95%) [33-35]. The mice were randomly assigned into five groups: sham-operated (PBS) group I, of which the uterine cavity was injected with 25 μL PBS and hold for 3 min; ethanol group II, for which 25 μL 95% ethanol was injected into the uterine cavity to induce damage for 3 min followed with PBS treatment; HA-treated group III, administration of HA after endometrium damage by 25 μL 95% ethanol as described above; HP-MSCs-treated group IV, administration of HP-MSC after endometrium damage by 25 μL 95% ethanol; and HP-MSCs-HA-treated group V, administration of HP-MSCs-HA after endometrium damage by 25 μL 95% ethanol. After acclimation for 7 days, the mice were sacrificed for further analyses. Figure S1 showed the construction and treatment procedure of the endometrium-injured mouse model in eight steps, which were briefly described as followings: I: mouse anesthesia; II: shaving the back of mouse; III: disinfecting exposed areas; IV: uteri exposure; V: instilling 25 μL ethanol in uterine cavity and holding 3 min to fully establish the model of thin endometrium; VI: intrauterine instillation of treating materials; VII: muscle suture; and VIII: closure of back skin incision.
Histological analysis and immunohistochemistry
Standard H&E staining was used for murine endometrial assessment. 30 treated female mice (6 mice, 12 uteri in each group) were euthanized on day 7 after surgery. The isolated uteri were embedded in paraffin after fixing with 4% paraformaldehyde overnight. The wax blocks were cut into 3-4 μm thick and stained with Hematoxylin-Eosin staining by standard methods to observe the endometrial thickness and the number of glands. Light microscopy photographs (OLYMPUS VS200, Japan) and endometrial images were analyzed using the Application Program Image Pro-Plus (version 6.0). Endometrial area and perimeter were recorded to assess an average measurement of the endometrial thickness (mean endometrial thickness = area/perimeter). Tissue sections were also labeled with Masson’s trichrome to measure the degree of endometrial fibrosis by conventional methods. The area of fibrosis was quantified by measuring the area ratio between endometrial stromal fibrosis and the endometrial area using a quantitative image analysis system (Image-Pro Plus software; Media Cybernetics, Bethesda, MD). The immunofluorescence staining was conducted as previously described [31]. Sections were incubated with primary antibodies Ki67, a rabbit polyclonal primary antibody (ab16667; 1:200; Abcam, Cambridge, UK). The secondary antibody (GK600711; DakoCytomation, Glostrup, Denmark) was applied for 30 min at room temperature. The number of positive staining cells in the glands and stroma was semi-quantitatively scored by the immune response score (IRS) of two observers who did not know the source of the samples. The scoring criteria were the same as previously described [31].
Fertility assessment
40 treated female mice (8 mice in each group) were mated with fertile males of the same age (female: male = 1:1) 7 days after surgery. The female mice were checked for vaginal plugs the next morning to determine whether pregnancy occurred. On the seventh day after the initial detection of vaginal plugs, the mice were sacrificed and the location and number of embryo implantation were recorded by photography.
Statistical analysis
All results were presented as mean ± SEM. We compared the means of samples using student's t-test between two groups and one-way analysis of variance (ANOVA) test among multiple groups. P-value < 0.05 was considered statistically significant.
{kind=link}