Animals
All mouse experiments were approved by the Animal Care and Use Committee at Zhengzhou University. C57BL/6 mice, Cx3cr1GFP mice (JAX # 005582), and Ai96(RCL-GCaMP6s) (JAX # 028866) mice were obtained from Jackson Labs, Ltd. Cx3cr1CreER mice were kindly provided by Dr. Steffen Jung from Weizmann Institute of Science. Cx3cr1GFP and Cx3Cr1CreER mice were maintained as heterozygous mice. Ai96 (RCL-GCaMP6s) were maintained as homozygous. Cx3Cr1CreER: RCL-GCaMP6s were obtained by crossing Cx3Cr1CreER heterozygous mice with Ai96 (RCL-GCaMP6s) homozygous mice and screening Cx3Cr1CreER heterozygous offsprings by PCR genotyping. Tamoxifen (75 mg/kg) was given intraperitoneally for 5 consecutive days at the age of two months to activate CreERT2 recombinase, which can excise the stop cassette in the genomic DNA of Cx3Cr1CreER: RCL-GCaMP6s mice and thus allow the transcription of downstream GCaMP6s. LPS from Escherichia coli O127:B8 (Sigma, L5024) was injected intraperitoneally (5 µg/g body weight) and brain tissue was collected 16 hours after injection. Both males and female were used and balanced numbers of sexes were mixed in our analysis and reports.
Primary microglia isolation
Primary mouse microglia were isolated as previously described with slight modification [15]. In brief, 7 to 9 weeks old mice were euthanized and transcardially perfused with ice-cold 0.0356% heparin sodium solution. The brain was removed and placed in an EP tube containing ice-cold 1X PBS. The whole brain was cut into pieces with an ophthalmic scissor, passed through a 22 gauge needle 5 times, and filtered with 70-μm filters. Brain homogenate was applied to a percoll gradient, and after a 30 min spin at 500 g, cells were collected from the 30%–70% interphase, pelleted, and washed.
Flow cytometry
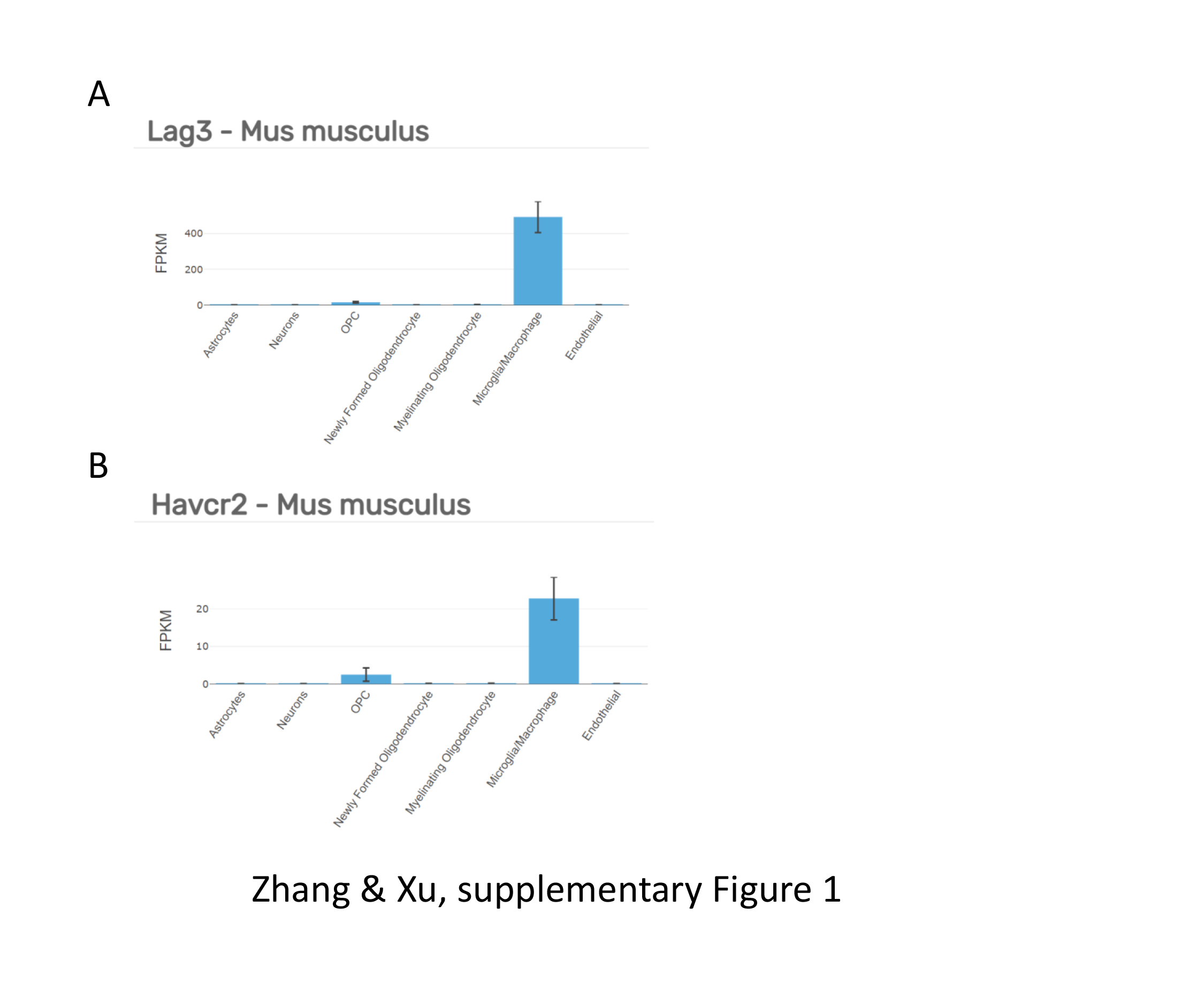
Several strategies were used to block the suspected binding of antibodies to Fcγ, including CD16/CD32 antibody (clone 93, Biolegend), CD16.2 antibody (clone 9E9, Biolegend), and mouse IgG (Solarbio). For antibody labeling, cells in the 200 µL ice-cold 1× PBS were stained with CD11b (clone M1/70, BD Biosciences), CD45 (clone30-F11, Biolegend) and LAG-3 (clone C9B7W, Biolegend) or TIM-3 antibodies (clone RMT3-23, Biolegend) for 30 min on ice. Flow cytometry analysis was performed on a BD FACScanto. Data were analyzed using FlowJo software (TreeStar).
Confocal imaging on fixed brain slices
Male and female mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA) in PBS, pH 7.4. Brains were post-fixed overnight in 4% PFA buffer, followed by cryoprotection in 30% sucrose in PBS for at least 48 h. Mouse brains were then embedded in Neg-50 frozen section medium (Fisher Scientific), sectioned using a cryostat at 40 µm, and mounted on coverslips for confocal imaging.
For immunostaining for fluorescence microscopy, sections were washed 3 times in 0.1 M PBS for 10 minutes each, before being incubated in a blocking solution containing 5% NGS in 0.1 M PBS with 0.5% Triton X-100 for 1 hour at room temperature on a shaker. For pretreatment of slices by lipofuscin quencher, slices were dipped in 1X TrueBlack® in 70% ethanol for 30 seconds. Sections were then incubated in primary antibodies (Iba-1, #019-19741, Wako; CD68, clone #FA-11, Biolegend) diluted in 0.1 M PBS with 0.5% Triton X-100 overnight at 4°C. The next day, the sections were washed 3 times in 0.1 M PBS for 10 min each before incubation at room temperature for 2 hours with alexa dye conjugated secondary antibodies (1:500; Invitrogen) diluted in 5% NGS in 0.1 M PBS. The sections were then rinsed 3 times in 0.1 M PBS for 10 minutes each before being mounted on microscope slides.
Confocal fluorescence images were taken using Plan Apo 60× 1.4 NA oil-immersion objective lens and the Nikon A1 confocal laser-scanning microscope. We used the 488 nm laser to excite GFP, with the intensity adjusted to 1% of the maximum output. The emitted light pathway consisted of an emission band pass filter (500–550 nm) before the photomultiplier tube. Autofluorescence was excited by the 561 nm laser line at 1% of the maximum output. The emitted light pathway consisted of a 570–620 nm emission filter. The spectral images were taken by an A1-DUVB-2 GaAsP detector unit (400-720 nm, 10 nm per step).
Brain slice preparation
For brain slice preparation, mice were deeply anesthetized with isoflurane and decapitated. Coronal brain slices (300 μm thickness) were prepared in chilled cutting solution comprising the following (in mM): 110 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 7 MgCl2, 25 d-glucose, 75 sucrose bubbled with 95% O2/5% CO2. During incubation, the slices were submerged at room temperature in aCSF comprising the following (in mM): 126 NaCl, 25 NaHCO3, 10 d-glucose, 2.5 KCl, 1.3 MgCl2, 2.4 CaCl2, and 1.24 NaH2PO4, bubbled with 95% O2/5% CO2. Brain slices were incubated at room temperature throughout the day. Slices were transferred to a recording chamber perfused with aCSF at a rate of 1–2 ml/min at 33°C.
Cranial window surgery
7 to 9 weeks old mice were implanted with a chronic cranial window. Briefly, mice were shaved head hair and injected with carprofen (20 mg/kg). During surgery, mice were anesthetized with isoflurane (5% for induction; 1–2% for maintenance) and placed on a heating pad. Using a dental drill, a circular craniotomy of >3 mm diameter; the craniotomy center was around the limb/trunk region of the somatosensory cortex. A 70% ethanol-sterilized 3 mm glass coverslip was placed inside the craniotomy. A dental Resin cement (3M-U200) was applied and air dried. A dental resin cement was applied to the rest of the skull, except for the region with the window. The dental glue was used to attach a custom-made head plate onto the dental resin cement of the skull. Mice were allowed to recover from the cranial window surgery for 2–4 weeks before the commencement of chronic imaging. Only surviving mice with a clear glass window were used for the imaging studies.
2-photon imaging on live brain slices and in vivo
2-photon fluorescence images were taken using the Nikon NIR Apo 40× 0.8NA water immersion objective lens and the Nikon A1 multi-photon microscope. On live brain slices, we used lock phased Coherent Chaemeleon 2-photon laser at 920 nm to excite GCaMP6s, with the intensity adjusted to 10% of the maximum output. The emitted light pathway consisted of an emission bandpass filter (505–525 nm) before the IR NDD. The spectral images were taken by an A1-DUVB-2 GaAsP detector unit (400-720 nm, 10 nm per step). In vivo, 820 nm laser was used to excite GFP and autofluorescence in microglia from Cx3cr1GFP mice. The emission light of GFP was passed through a 500-550 nm band pass filter and autofluorescence through a 600-656 nm band pass filter.
Quantification and statistical analysis
Sample sizes were based on similar previously published work. The results of statistical comparisons, n numbers and p values are shown in the figure panels or figure legends with the average data. All statistical tests were run in GraphPad Prism 8. The graphs were created in GraphPad Prism 8 or Origin 8 and assembled in Powerpoint 2016. No data has been excluded from the analysis. Since the mice used in our study are all in congenic C57BL/6J background and thus no cofounder would be expected to affect the comparison between different treatment groups, therefore, animal selection has not been randomized. Since no subjective methods have been used in our study and thus no bias during data collection would be expected, investigators were not blind to the groups during data collection. The normality of the data distribution was determined using the Shapiro–Wilk test before appropriate statistical methods were chosen. If the data were normally distributed, two tailed Student’s t test or ANOVA were used. If data were not normally distributed, non-parametric Mann-Whitney test was used.
{kind=link}