Animals
All animal procedures were carried out in accordance with the guidelines provided by the Institutional Animal Care and Use Committee of the University of Chicago (Chicago, Illinois). The C57BL/6-MMP3 deficient (MMP3−/−) mice were a GEM Collection Model TTM-610 and were generated as described before [19]. The line was backcrossed for 12 generations with C57BL/6 mice, which were provided by the Taconic Biosciences company. A PCR-based analysis was employed to genotype mice. Mice were housed in a room at 22-24°C on a 12-hour light/dark cycle and received drinking water ad libitum. We used 8-week-old male mice in this study.
Genotyping: We isolated genomic DNA from mouse tail clips using the Puregene DNA isolation kit from Gentra Systems according to the manufacturer’s instruction. About 10ng of the genomic DNA was used for PCR.
Experimental Design
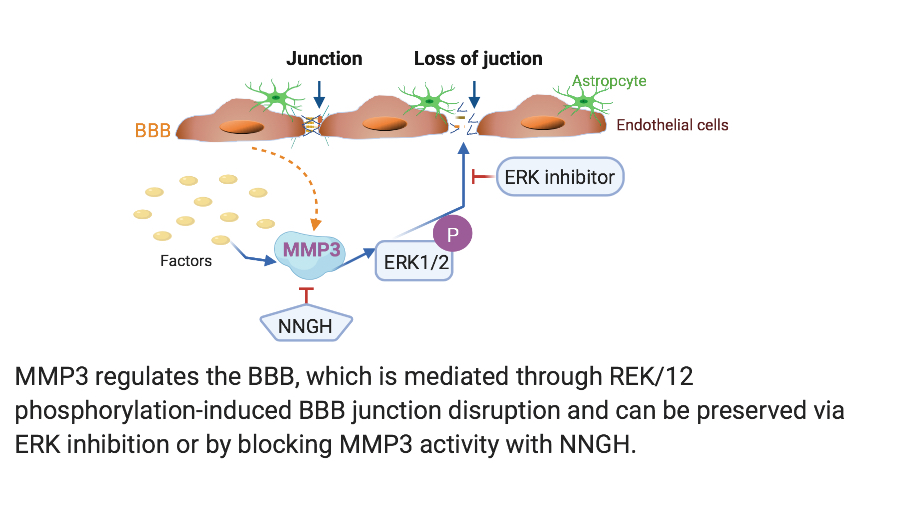
The experimental design of the current study is described below and illustrated in Figure 1. To investigate the influence of MMP3 on the BBB integrity, we assigned all the mice to three different groups under isoflurane, including WT (MMP3+/+), KO (MMP3-/-), and WT+MMP3 (the mice were administered a recombinant human MMP3 via tail intravenous injection (iv), which was purchased from Abcam). Each experimental group contained 8 mice. In vivo, the BBB permeability was evaluated by measuring Evans blue and sodium-FITC-dextrans extravasation [20]. In vitro study, the BBB permeability was detected in an in vitro model of BBB. To evaluate the role of MMP3 on BBB junction proteins, we used the MMP3 recombinant human protein to stimulate brain microvascular endothelial cells (BMVECs). Afterward, we detected the protein level of MMP3, ZO-1, occludin, VE-cadherin, and claudin-5 by western blot and fluorescence staining. To increase the MMP3 levels, lipopolysaccharide (LPS) in a dose of 100ug/ml as a stimulus to increase the MMP3 levels [21, 22], and then the ERK inhibitor FR1080204 [23] (purchased from Sigma-Aldrich) was administered to investigate the mechanism behind the effects of MMP3 on the BBB.
In Vitro Model of the Blood-Brain Barrier
To mimic the anatomic structure of the in vivo BBB, we developed an in vitro BBB model based on the co-culture of brain ECs with astrocytes, as described in other publications [24]. In this model, BMVECs were seeded in the transwell insert, and then primary isolated astrocytes were grown on the undersurface of the transwell insert. Isolation of the BMVECs and astrocytes are described in detail in the supplemental file.
MMP3 Administration In Vivo and In Vitro Study
The MMP3 recombinant human protein purchased from Sigma was diluted to 20µg/ml in the assay buffer: 50mM Tris, 10mM CaCl2, 150mM NaCl, 0.05% (w/v), and Brij-35, pH 7.5. For the next step, MMP3 was then activated by adding chymotrypsin (Sigma, 1mg/ml stock in 1mM HCl) to a final concentration of 5ug/ml. Afterward, the preparation was incubated at 37°C for 30 minutes [25], and a dose of 50ug/kg of the MMP3 preparation was injected into the tail vein 30 minutes before an Evans blue injection in vivo. The activated MMP3 was added to the in vitro cells at a dose of 150ng/ml for 24 hours.
Isoflurane Exposure
We anesthetized the mice with isoflurane in the chamber until they lost consciousness and respiration showed to an appropriate rate. A typical exposure includes 3% isoflurane and 1% O2, which corresponds to a respiration rate of 1 breath every 2 sec. A response to a toe pinch or tail pinch was used to confirm the appropriate state of anesthesia. The mice were orally intubated with a 22G IV catheter and artificially ventilated with a rodent respirator (Harvard 1. Apparatus or similar device, tidal volume 0.3ml, rate 105 strokes/min) [26]. The air from the thorax was evacuated through a tub, which was connected from the mice noses and extended to a Fluosorber canister. Anesthesia was maintained with 1–2% isoflurane in 100% oxygen for 1 hour. The concentration of isoflurane was adjusted according to the loss and regain of reflexes. During anesthesia, mice were placed on a heating pad to maintain body temperature at 37°C and monitored until sternal and fully ambulatory. Anesthesia was terminated by discontinuing isoflurane administration. The anesthetic effect of isoflurane (induction time, emergence time, and isoflurane usage volume) was recorded carefully.
In Vivo BBB Permeability Assay in Mice
Either Evans blue [27] or a sodium-Fluorescein Isothiocyanate (FITC) assay [28] were used for the assessment of the BBB integrity. The mice were injected with 2% of Evans blue or sodium-FITC (1mg/ml in saline) 200ml through the tail vein 2 hours before sacrificing them. Moreover, they were anesthetized with 1-2% isoflurane for 1 hour and then perfused with 10 ml normal saline to remove the Evans blue dye and the sodium-FITC from the blood vessels. Brain tissue from the bilateral temporal lobes was carefully removed from the animals following sacrifice. Regarding the mice injected with Evans blue dye, a total of 0.5ml of formamide was added to the brain tissue homogenate to dissolve the Evans blue dye. After incubation in a 55°C water bath for 48 hours, the samples were centrifuged at 12000 × g for 30 minutes. The supernatant was then collected before absorbance measurements at 632nm were performed using a UV spectrophotometer (Hitachi, Ltd.). The Evans blue content was calculated according to the standard curve. For mice that were injected with sodium-FITC, a total of 1ml 50mM of Tris buffer solution was added to the brain tissue homogenate and then centrifuged at 3000rpm for 30 minutes. We proceeded to collect the supernatant, which was then mixed with methanol (1:1) and centrifuged at 3000rpm for 30min. The supernatant was collected, and its fluorescent intensity measured on a plate reader. Finally, the concentration per mg of tissue was calculated using a standard curve.
RNA Isolation, Reverse Transcription, and Quantitative PCR
Total RNA was isolated from cells and tissue using a RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. The extracted RNA was treated with recombinant DNAse (Invitrogen, Grand Island, NY) according to the manufacturer’s protocol to remove any DNA contamination. cDNA was synthesized from 500ng of RNA using the Superscript III reverse transcription kit (Invitrogen). Quantitative real-time PCR was performed using a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA) to determine the gene expression for MMP3 using validated TaqMan® gene expression assay primer/probe combinations (Applied Biosystems). All qPCR results were normalized to the expression of the endogenous control 18S. Folding changes in the transcripts were determined using the delta cycle threshold (i.e., ΔΔCt) method.
Transendothelial electrical resistance (TEER) measurement
Measurements of transendothelial electrical resistance (TEER) across the mice brain EC monolayers were performed using the electrical cell-substrate impedance sensing system (ECIS) (Applied BioPhysics, Troy, NY, USA), as described in a previous study. [29] [30] For impedance measurements, cells were grown on 8W10E+ arrays. The arrays were treated with 10 mM L-cysteine (cat#C7352-25G, Sigma-Aldrich) followed by coating with collagen type I (cat#A1048301, Thermo Fisher Scientific) at 1μg/cm2. The arrays of the electrical stabilization command in the ECIS software were used to sterilize and clean the gold electrodes. BMVECs were seeded onto the arrays at a density of 60,000 cells/cm2 in 400 μL of L-DMEM growth media. ECIS was conducted using the multiple frequency/time (MFT) option to record the impedance measurements over a broad spectrum of frequencies.
Detection of BBB Permeability In Vitro Study
To construct an in vitro BBB model [31], we isolated and identified mice BMVECs and astrocytes (Supplementary Figure 1). We placed a Transwell chamber insert with a 0.4-μm aperture (Corning, New York, USA) into volumetric flasks. Astrocytes (1X106/mL) were added to the underside of the Transwell and incubated at 37°C and 5% CO2 for 24 hours. The chambers were then placed carefully into a 6-well plate (Corning) (Figure 1B). When the astrocytes reached 60% confluence under an inverted microscope, ECs (1X107/mL) were seeded on top of the Transwell (Figure 1C) at 37°C and 5% CO2. Furthermore, we observed the cells under an inverted microscope until the astrocytes and BMVECs reached a high-density co-culture and, at that point, a FITC-dextran (Dextran-blue-3KD and Dextran-red-40KD) [32] transwell assay was used to assess the in vitro BBB permeability.
Western Blot
Cells were placed on ice, and the media was aspirated prior to washing twice with ice-cold PBS. A cell lysis buffer was prepared by adding a complete mini protease inhibitor tablet (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitors (Sigma-Aldrich) to a protein isolation buffer (40mM HEPES, 120mM NaCl, 1mM EDTA, 3% CHAPS w/v). Ice-cold buffer was added to each well, and the cells were mechanically disrupted using a rubber scraper. We performed western blots, as described before in my paper [33]. Briefly, protein extracts from the cells were subjected to a freeze/thaw cycle before being centrifuged (6,000rpm × 20 min). Supernatants were collected, protein concentrations were determined using a BCA assay (Thermo Fisher SDS-PAGE was performed using 4–12% Bis/Tris gels (Biorad), and proteins were transferred to nitrocellulose membranes. After blocking with 5% non-fat milk in TBST, we incubated the membrane with anti-MMP3 (1:1000; Abcam), anti-ZO-1 (1:1000, Invitrogen), anti-occludin (1:1000, Invitrogen), anti-VE-cadherin (1:500, Invitrogen), anti-claudin-5 (1:1000, Invitrogen), and anti-p-ERK (1:2000 Invitrogen) antibodies for 16 hours at 4°C. After incubating with a Horseradish peroxidase (HRP)-conjugated secondary antibody, the membranes were developed with enhanced chemiluminescence (ECL) reagent and imaged with a ChemiDoc imaging system (Biorad). A quantitative assessment was performed using Image Lab software (Biorad).
Assessment of Protein Expression by Quantitative Immunofluorescence
Fixed samples were thawed and rehydrated with PBS containing glycine at 50mM (Sigma-Aldrich) for 10 minutes. Cells were then permeabilized with 0.1% Triton X-100, and non-specific binding sites were blocked with a blocking buffer (PBS containing 1% bovine serum albumin BSA and 10% serum from the species the secondary antibodies were raised in). Cells were incubated overnight at 4°C with primary antibodies against one of the following targets: vWF (1:500 dilution; Dako, Carpentaria, CA), GFAP (1:50 dilution; Invitrogen, Carlsbad, CA), MMP3 (1:50 dilution; Invitrogen, Carlsbad, CA), or ZO-1 (1:50 dilution; Invitrogen, Carlsbad, CA). After incubation with the primary antibodies, the slides were washed and incubated with fluorescent-labeled secondary antibodies (1:200 dilution; Invitrogen, Carlsbad, CA), and mounted under glass coverslips with Vectashield-containing DAPI for nuclear identification (Vector Laboratories, Burlingame, CA). Slides were imaged on a fluorescent microscope and analyzed using Image J software. The average maximum intensity was calculated, and this value was used as the measurement threshold. Next, ten fields were selected at random, and the average of the maximum and mean intensities were calculated correcting for the background threshold. Three images per animal were assessed.
Detection of MMP3 Levels
Both MMP3 serum levels and MMP3 protein expression levels in cultured BMVECs were also determined using a specific ELISA kit by following the manufacturer’s instructions (ELISA kit Abcam; ab203363). 50μl of serum (diluted 100 times) and cell culture supernatants or standards were added together with an antibody cocktail to appropriate wells. After incubating for 1 hour at room temperature on a plate shaker set to 400rpm, the microplate was washed 3 times. Further, 100μl of TMB Substrate was added to each well. Finally, the OD was recorded at 450nm after stopping the reaction by adding 100μl of Stop Solution.
Statistical Analysis
All data are shown as the mean ± SD. Data were determined to be normal by the Shapiro-Wilks test, and unpaired t-tests were used for comparisons between two groups. For comparisons between multiple groups, a one-way ANOVA with Tukey’s multiple comparisons test or a two-way ANOVA with Bonferroni post hoc test were performed depending on the number of experimental variables. A two-tailed p-value of less than 0.05 was considered statistically significant. Data were visualized and analyzed using SPSS version 17.0 (Armonk, NY) and GraphPad Prism version 8.0.0 (San Diego, CA).
{kind=link}