Drug discovery and development is one of the most important components of the pharmaceutical industry. In order to meet the customers’ increasing demands while guaranteeing the good potency and minimal side effects, structures of new drug molecules have been becoming more and more complicated. In other words, extensive pharma R&D activities are dramatically required, and relative short discovery-development-deployment cycle is desirable[1].
Over the past decades, as a rate-limiting factor, innovations in organic synthesis have significantly enabled the discovery and development of important life-changing medicines, improving the health of patients worldwide[2, 3]. Nevertheless, innovations and excellence in organic synthesis are expected to be the most powerful driver for all phases of drug discovery and development. Very recently, chemists have enthusiastically applied advanced machine learning and artificial intelligence (AI) technologies towards the preparation of drug molecules. For example, AI driven discovery of drug molecules[4-6], automated planning of synthetic routes[7-9], machine learning driven optimization of reaction conditions[10-12], and autonomous assembly of synthetic processes[13-15].
Synthesis planning, which is regarded as one of the central elements of organic synthesis, can be traced back to 1960s[16]. Traditional computer-aided approaches for synthesis planning are labeled with different disadvantages such as low efficiency, poor repeatability and high experimental cost. Especially, more and more compounds and reactions have been discovered. For example, the number of reactions and compounds that Reaxys database contained has exceeded 40 million and 100 million, respectively. Novel synthesis planning approaches are highly required. During the past 3 years, a variety of machine learning and artificial intelligence methods such as random forest, automated reasoning, support vector machines and more recently deep learning have demonstrated their capacity for organic molecules discovery, design, and production[13, 17-19]. Clearly, the application of the aforementioned new technologies to end-to-end organic molecules discovery and development will be the key point for realizing the fully automated synthesis planning[20].
Retrosynthetic analysis is the canonical technique used to plan the synthesis of small organic molecules for drug discovery[21]. In General, retrosynthesis analysis consists of four steps: step 1. determine the target compound; step 2. disconnect certain bonds which are thought to be easy to form according to known chemical knowledge in the target compound as the reverse of reaction, and search for possible precursors in this way; step 3. repeat step 2 for all the precursors to form a synthesis tree, and expand it until all precursors are available; step 4. evaluate all the branches of the synthetic tree one by one, then take the most possible one as the optimal route. Since different groups of reaction sites in the same group of precursors may exist, in other words, the difference between real reactions and the expected ones in the branches may occur, step 4 is therefore very critical but also the most difficult one. Forward reaction prediction is a necessity point to guarantee the correct evaluation of each branch. The main aim of the paper is to present the machine learning approaches for assisting the forward reaction prediction.
The existing approaches for forward reaction prediction can be categorized as template-based and template-free method. For example, Coley et al.[17] applied reaction templates to reactants to generate candidate reactions as many as possible, which then are used to train a neural network. While for template-free methods, Schwaller et al.[22] compared chemical reactions from reactants to products to translations from one language to another, so that forward reaction prediction can be transformed into machine translation and solved by training a seq-to-seq recurrent neural network which directly takes reactant SMILES (Simplified Molecular Input Line Entry Specification) as input.
As the research target is to discover new reactions meaningful for organic synthesis according to existing reaction mechanisms, to fully reuse the discovered reaction rules summarized from experimental results so far, the presented paper adopts the template-based approach. Specifically, the concentration of this paper is to adopt a novel hard-threshold based deep neural network for improve the prediction accuracy for the forward reaction prediction. In detail, hard-threshold activation and target propagation algorithm are implemented by introducing the mixed-convex combinatorial optimization. Comparative tests are conducted to explore the optimal hyperparameter set. The reminder of the paper is as follows: forward reaction prediction will be described firstly, then hard-threshold neural network is touched upon. Results and discussions are then given. Conclusion is finally presented.
Forward reaction prediction
The above reaction can be decomposed into three Edits: atom 1(nitrogen) loses a hydrogen, atom 2(carbon) and 3(chlorine) loses a single bond, and atom 1 and 3 respectively gains a single bond. In the reactant, atom 1 has one hydrogen and two non-hydrogen neighbors, atom 2 has no hydrogen but is surrounded by three non-hydrogen neighbors, while atom 3 has one non-hydrogen neighbor without any hydrogen. Therefore, feature vectors of atom 1, atom 2, atom 3, along with two bonds can be expressed as: (see formula set 1 in the Supplementary files)
The Edit Vectors of hydrogen loss and gain (e1, and e2, respectively) are directly taken as the feature vector of the corresponding atom, and those of the bond loss and gain (e3, and e4, respectively) are taken as the connection of the feature vectors of corresponding atoms and the bonds: (see formula set 2 in the Supplementary Files)
Where "/" represents the connection of feature vectors, and the outermost brackets represent that Edit Vector is the combination of all the vectors of basic Edit in a reaction. For instance, if one reaction has four atoms losing hydrogen in the reactant, its Edit Vector of hydrogen loss (e1) will have four 6D feature vectors.
Candidate reaction selection
The selection step uses a complex neural network consisting of several subnetworks, as shown in Fig.3. For each candidate reaction, the above mentioned four Edit Vectors are calculated and severed as input to four corresponding subnetworks. The sum of outputs from four subnetworks is then fed to the lower integrating subnetwork to produce scalar probability scores. The above steps are repeated for all candidate reactions, and then all the probability scores are normalized by the softmax method to estimate the probability of occurrence of each candidate reaction. Finally, all candidate reactions are sorted by the probability score, and the candidate ranking first is regarded as the prediction result. The prediction is right if its outcome has the same SMILES as the recorded one, and vice versa.
In order to improve the prediction accuracy, a hybrid model using the Edit Vector and the Extended-Connectivity Fingerprint (ECFP)[23] is also considered in our work. The only difference between two models is that an extra subnetwork without hidden layer which evaluates the ECFP is added to the hybrid model, as shown in the middle of Fig.3. The output of ECFP subnetwork is multiplied by ε when subnetwork outputs are summed before input to the final integrating subnetwork, where ε is the mixing factor. By adjusting ε, the proportion of ECFP subnetwork can be precisely controlled.
Hard-threshold neural network
Neural network, especially deep neural network learning, is currently the most popular machine learning algorithm with powerful fitting capability[24]. However, with the ceaseless expansion of the size of the neural network, a series of problems, particularly gradient vanishing and gradient explosion would often occur. To get rid of this dilemma, the paper tends to apply the hard-threshold neural network for predicting the outcomes of organic synthesis.
Constructing hard-threshold neural network
“Hard-threshold neural network” means neural network with hard-threshold activation, including Step activation and Staircase activation, which is shown in Figs 4a and 4b, respectively. Indeed, staircase activation is the sum of some step activations. Actually, hard-threshold activation was used in the early contributions to deal with the binary classification when the neural network had not been proposed. Hard-threshold activation has a constant derivative as 0, which can effectively avoid gradient vanishing and gradient explosion. Besides, the scale of output is almost fixed and insensitive to the scale of the input, which helps avoid certain abnormal propagation and simplify the computation. However, the zero derivative of hard-threshold activation also prevents it from being trained with traditional backpropagation.
Target propagation algorithm
A new backpropagation algorithm is required to train a hard-threshold neural network bypassing the zero derivative of hard-threshold activation.
Based on the “target propagation” concept[25], a new target propagation algorithm named FTPROP-MB was recently proposed[26]. Basically, since perceptron with Step activation is trainable, hard-threshold neural network could also be trainable if it can be decomposed into perceptron. Specifically, a target vector td is introduced to represent what the dth layer is supposed to be the output for all hard-threshold activation layers. After the normal forward propagation procedure for each layer, FTPROP-MB determines td firstly, then introduces a layer loss Ld which is used to compute gradient just like training perceptron so that weights can be updated.
Considering that the output of the hard-threshold activation is a set of discrete values, the determination of td can be reduced to a combinatorial optimization problem. In detail, the question of how to optimize td regarding overall loss and layer loss can be expressed in standard form as follows: (see Equation 1 in the Supplementary Files)
where Wd and zd represents the weight and the pre-activation output of the dth layer. The search space is large and discrete because all the components of td is restricted to 0 and 1, so it is tough for common search algorithms to find the optimal solution in reasonable time horizon. Since layer loss is usually convex, FTPROP-MB determines the target vector td in the dth layer with a heuristic method: compute the derivative of layer loss in the (d+1)th layer Ld+1 with respect to the dth layer’s output hd, and then set td according to the opposite sign of this derivative. This method can be mathematically formulated as:(see Equation 2 in the Supplementary Files)
When layer loss function is convex, the negative partial derivative of Ld+1 on hdj points to the global minimal of Ld+1. Let’s take hdj=-1 as an example, if r(hdj)=-1, which indicate that the partial derivative of Ld+1 is positive, it is clear that Ld+1 will increase by making hdj=+1 when fixing other components of hd, so there is no doubt that tdj=r(hdj)=-1. On the other hand, when r(hdj)=+1, which means the partial derivative of Ld+1 is negative, we don’t know exactly whether Ld+1 would increase or decrease by making hdj=+1. But the difference between hdj and r(hdj) indicates that the current value of hdj is lack of confidence, so a natural choice is to lead zdj to 0 by adjusting tdj to make it more possible for hdj to flip, w.r.t tdj=r(hdj)=+1.
In summary, the training process of a n-layer hard-threshold neural network has both optimization problem on weights and convex-combinatorial optimization problem on target vectors, so a mixed convex-combinatorial optimization problem is formed. The block diagram of target propagation algorithm is shown in Fig.5.
Layer loss function
Till now we are still facing a problem of choosing layer loss function. According to related work[27], it is acceptable to adopt soft hinge loss and weighing according to the gradient, which is shown in Fig.6.
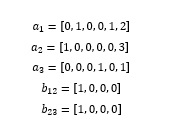{kind=link}
{kind=link}
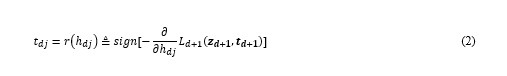{kind=link}
{kind=link}