Cells and Antibodies
Chicken embryo fibroblast cell lines, DF-1 cells, were obtained from ATCC (Manassas, USA) and were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 0.1% penicillin/streptomycin at 37 °C in an atmosphere of 5% CO2. The ALV-J envelope protein that is a specific mouse anti-monoclonal antibody, JE9, was kindly provided by Prof. Aijian Qin, Yangzhou University. Goat anti-mouse IgG labeled with FITC was purchased from Bioss (China), while the ALV antigen-capture enzyme-linked immunosorbent assay kit was purchased from IDEXX (USA).
Animals
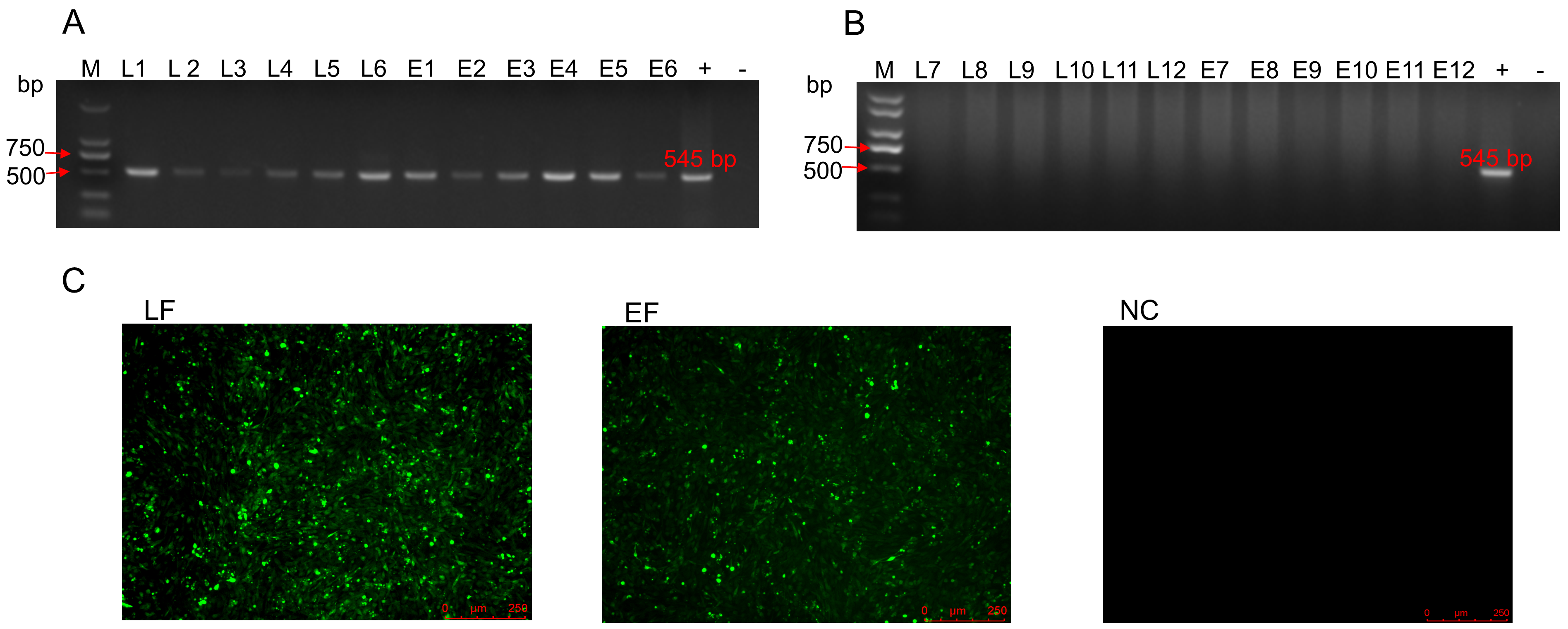
The 140 d LF and EF yellow chickens were sourced from Chinese chicken farms in Guangdong Province, China. Virus isolation and identification were performed in DF-1 cells as we described previously [20]. The virus identification primer sequences are listed in Table S1. The virus identification and ALV-J viremia results are shown in Fig. 1S and Table S2.
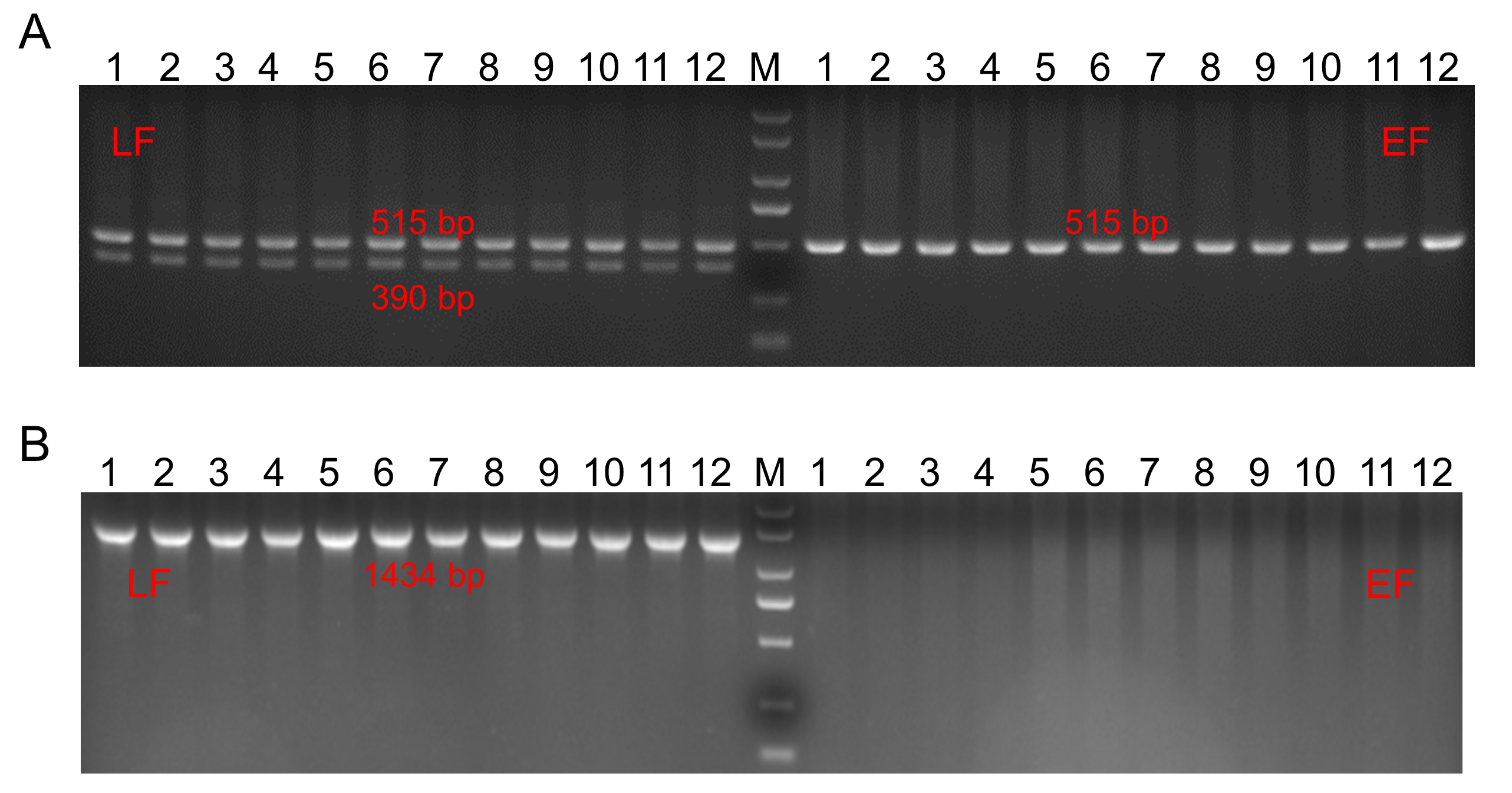
We used molecular identification methods to identify the feathering genotypes of the samples. Using primers designed by Tixier-Boichard et al. [21]. we amplified the ev21 gene in our DNA samples. The expected PCR product of EF chickens is only a 515 bp band, while for LF chickens it is two bands of 390 and 515 bp. We designed a pair of primers for dSPEF2/dPRLR gene amplification. A 1434 bp target fragment should be obtained in LF chickens but not in EF chickens. The primers are listed in Table S1. The feathering genotype detection results of LF and EF chickens are shown in Fig. 2S.
Based on the results of virus isolation and identification and the feathering genotype detection, 6 ALV-J positive LF chickens, 6 ALV-J negative LF chickens, 6 ALV-J positive EF chickens and 6 ALV-J negative EF chickens were selected. All animals were from the same farm. These selected chickens were raised separately, with a consistent feeding protocol between all individuals.
Sample collection
Once each week for eight weeks, we aseptically collected 2 mL of anticoagulated blood from each individual. The plasma was then separated by centrifugation at 2000 rpm at 4℃ for 15 min and stored at - 80℃. All the samples were collected at the same time: 10:00 am every Monday of each week. Eight weeks later, the spleen, bone marrow, thymus and cecal tonsil of each chickens were collected and stored at - 80℃. The plasma samples were analyzed with a p27 test for each collection, and the cells supernatant p27 test results were shown in Table S2.
Determining of the levels of PRL, GH, IgG and IgM in the serum by ELISA
The levels of the PRL in the serum was measured using enzyme-linked immunoassay (ELISA) kits for PRL (CLOUD-CLONE, Wuhan, China), following the manufacture’s protocol. The levels of GH, IgG and IgM were detected in the serum by specific ELISA kits purchased from Shanghai Enzyme-Linked Biotechnology Co. Ltd (Shanghai, China), following the manufacture’s protocol.
Cell’s infection and overexpression
The infection of DF-1 cell with ALV-J was carried out as our previously described [22]. After incubation for 24 h and 48 h in culture, we collected cells and extracted RNA, and then measured the PRLR and SPEF2 expression by qRT-PCR. The laboratory ALV-J strain SCAU-HN06 was kindly provided by Prof. Weisheng Cao (South China Agricultural University, Guangzhou, China).
According to the PRLR sequence (NM_204854.1) on the NCBI database, Wuhan Genecreate Industrial Co., Ltd was commissioned to construct the pcDNA3.1-PRLR and the pcDNA3.1-dPRLR plasmid. Then we followed the method described by Li et al.[22]. for cells transfection and cells infection. After incubation for 24 h and 48 h in culture, we collected cells and extracted RNA, and then measured the ALV-J virus expression by quantitative real-time polymerase chain reactions (qRT-PCR).
RNA isolation and cDNA synthesis
Total RNA was extracted from tissues with RNAiso reagent (Takara, Japan) according to the manufacturer’s protocol. The integrity and quantity of RNA were assessed using 1% agarose gel electrophoresis and spectrophotometry (ND-2000, USA), respectively. cDNA was synthesized using MonAmp™ RTIII All-in-One Mix (Monad Co., LTD Guangzhou, China), following the manufacture’s protocol. The synthesized cDNA was stored at -20℃ until subsequent analysis using qRT-PCR.
Quantitative real-time PCR
Immune-related differentially expressed genes, TLR4, TLR7, MDA5, SOCS3, VIP, IL-10, IRF1, NFkB, TNFα and IL-1β, were selected, and the expression of each gene was detected in the spleen of the LF and EF chickens. The MonAmp™ SYBR® Green qPCR Mix (Monad Co., LTD Guangzhou, China) was used for qRT-PCR in an ABI 7500 Real-Time Detection instrument (Applied Biosystems, USA) according to the manufacturer’s protocol. Relative gene expression was measured by applications of qRT-PCR for each sample and the nuclear gene GAPDH was used as a control. The primers used in the qRT-PCR are shown in Table S1.
Western Blotting Assay
Western Blotting (WB) Assay were performed as previously described [23]. The antibodies and their dilutions used for WB were as follows: anti-ALV-J envelope protein specific monoclonal antibody JE9 (kindly provided by Prof. Aijian Qin, Yangzhou University, 1:1000). Rabbit Anti-beta-Actin antibody (Boss, China; 1:500), Goat Anti-Rabbit IgG H&L/HRP antibody (Boss, China; 1:500).
Statistical analyses
Statistical comparisons were performed using GraphPad Prism 5 (GraphPad Software Inc., USA). The data were presented as means ± one standard error of the mean (SEM). The statistical analyses were performed using one-factor analysis of variance, and statistical significance was represented by P-values. P < 0.05 was considered statistically significant, and P-value bands of statistical significance were denoted as: *P < 0.05, **P < 0.01, ***P < 0.001.
{kind=link}
{kind=link}