Study Design
This is a parallel, two-arm, non-blinded, randomized controlled trial in the single center.
Study site
The study will be conducted in the city of Imperatriz, Maranhão in diabetic patients with chronic foot ulcers (classified as grade 2, 3 and 4 of Wagner [23] and persisting for more than one month). The outpatient follow-up for diabetic foot patients will be done at the SUS (Unified Health System - a public health service for the entire Brazilian population), with HBOT sessions being held in the CicatrizAR Clinic.
Sample and Study Period
The sample will consist of diabetic patients with chronic foot ulcers treated with standard methods, i.e. dressings, debridement, antibiotics, and load relief (control group), and to the other group will be added HBOT (HBOT group). The study will be conducted from 2019 to 2020.
The sample size has been calculated based on the formula for comparing two independent groups according to qualitative variables [24], with 95% confidence interval and 80% power. A wound healing rate of 90% was considered achievable for the case group at one year, which we expected to be at least 20% higher than in the control group (absolute difference, i.e. no more than a wound healing rate of 70% was expected in the control group). Thus, the total sample size was estimated as 60 patients per group (120 in total).
Trial Status
Registration number RBR-7bd3xy. Registered 17 July 2019. The first recruitment was held on 04 July 2019 and the last recruitment is foreseen on 31 August 2020.
Eligibility criteria
The inclusion criteria of the study are adult patients (age > 18 years), stable clinical presentation, diabetes type 1 and 2, Wagner Grade 2, 3, and 4 foot ulcers, ulcers persisting for more than one month without cure, authorisation for the study, and patients of the SUS (Unified Health System).
Exclusion criteria of the study are failure to meet one of the inclusion criteria, patients with macroangiopathy (two distal pulses absent), and patients with contraindications for HBOT, absolute or relative, patients on bleomycin chemotherapy, with chronic obstructive pulmonary disease, previous spontaneous pneumothorax, chronic sinusitis, chronic otitis media, unstable angina, severe congestive heart failure, claustrophobia, severe dementia, depression, and history of seizures.
Data collection
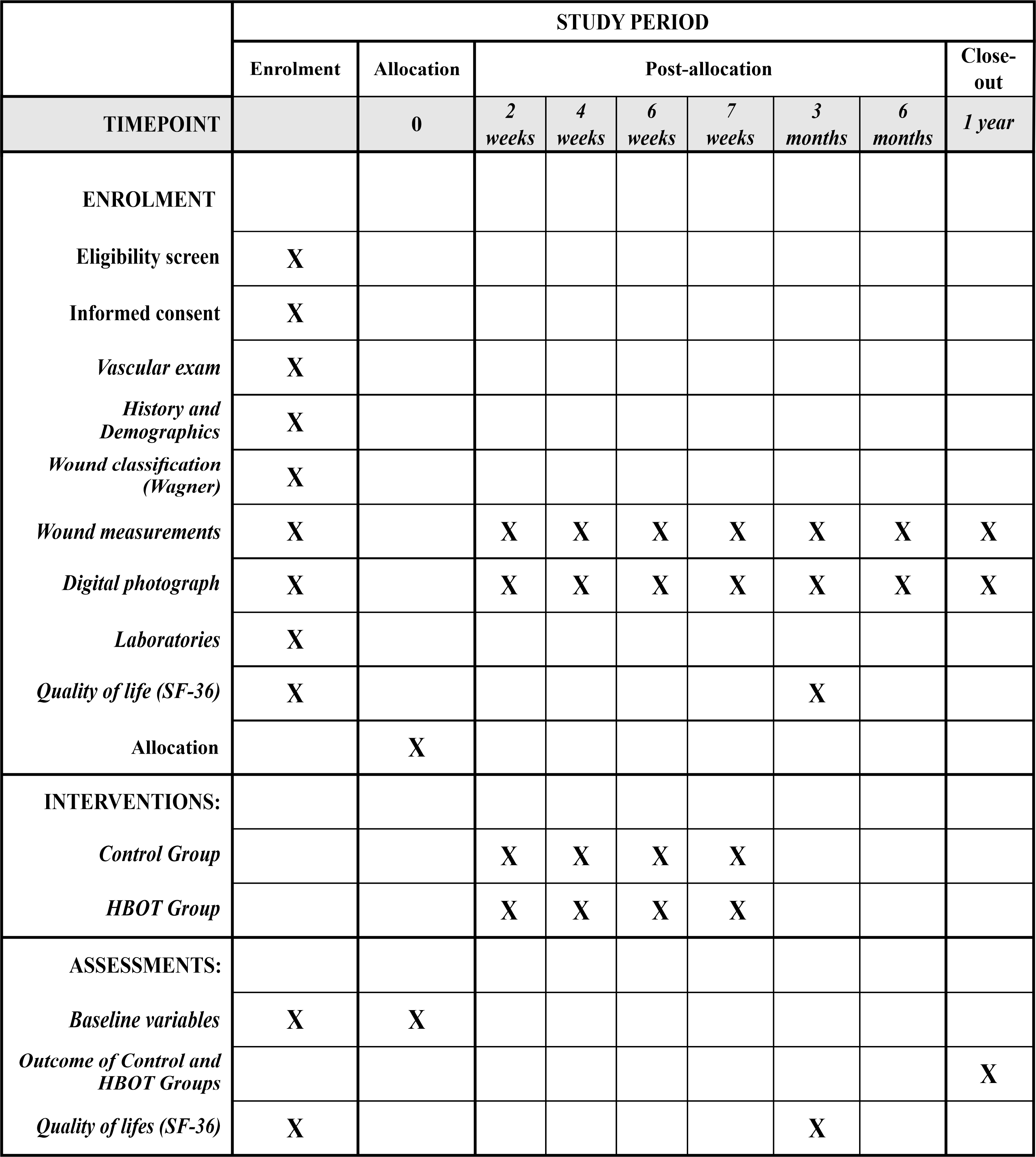
The data collection will be performed in the diabetic foot outpatient clinic of the SUS and in the CicatrizAR clinic, using a standardised questionnaire provided in the Supporting Information, as well as with photographic recording of the ulcers, according to the timeline described in Figure 1.
Figure1. The schedule of enrolment, interventions, and assessments.
Randomization
This is a parallel, two-arm, non-blinded, randomized controlled trial in which an randomization will be performed by the research coordinator, who will carry out by simple 1: 1 draw (for each pair of patients, one participant will select the number 1 or 2 in the draw, which will correspond to the group to be allocated, while the second patient will be allocated to the other group.). The randomization will not be done by computer generated random numbers. Patients will be selected at the SUS diabetic foot clinic, where they regularly monitor the lesions on their feet. They will be evaluated according to the inclusion criteria of our research and, if they meet the criteria, they will be invited to participe of the research. The researchers explain how the study will be carried out and after 1 week, if the patient is interested in participating in the study, the research coordinator collects the signature of the patient's consent form, that found in the support information for this article, as well as performs the randomization of patients to the research groups.
Interventions
The patients of the HBOT group will be evaluated upon admission, after 10, 20, 30 and 35 HBOT sessions and after six months and one year; and the control group will also be assessed at equivalent periods (upon admission, after 2, 4, 6 and 7 weeks, six months and one year) to clinically evaluate the ulcers and perform specific measures using the software ImageJ, developed at the National Institutes of Health (NIH, Bethesda, Maryland). HBOT sessions will be held 1 week after randomization. The sessions will be supervised by doctors trained in hyperbaric medicine by the Brazilian Society of Hyperbaric Medicine (SBMH). The HBOT sessions will be conducted five days a week, being carried out with patients in a multiplace chamber at 2.5 atmospheres absolute (ATA) and 100% O2, with 10 minutes for compression and decompression, effective sessions of 90 minutes, with intervals of 5 minutes for each 30 minutes of treatment. The dressings will be done in both groups with Exufiber foam Ag ® (Molnlycke, Gotemburgo, Suécia) or Mepilex® (Molnlycke, Gotemburgo, Suécia), which will be chosen according to the characteristics of wounds, with silver dressings for the infected injuries and a foam with polyurethane protective layer for wounds in which granulation tissue has formed. Secondary dressings will be changed daily and the primary dressings (coverings - Exufiber foam Ag or Mepilex) will be changed every 3-5 days, as needed and in both groups. The progression of the wounds and specific treatment, such as appropriate bandage, antibiotic therapy, or need of some surgical intervention will be evaluated on a weekly basis. In both groups, a vascular surgeon will monitor the patients, and who will assess the need for surgical procedures (debridement and amputations), in addition to appropriate antibiotic therapy for each case. With regard to load relief, patients in both groups will wear orthopedic Baruk shoes.
The SF-36 quality of life questionnaire will be filled upon admission and after three months of follow-up in both the groups. Laboratory examinations, such as a haemogram, erythrocyte sedimentation rate, levels of C-reactive protein, creatinine, fasting blood glucose, and glycosylated haemoglobin will also be estimated only on admission.
Primary outcome
Wound healing will be assessed by evaluating the diameter of the lesions by specific software and periods, as described in the interventions. The wound healing will be achieved when there is no more skin lesion on the skin and the primary endpoint will be a binary result. The assessor of the primary outcome will be blinded.
Secondary outcome
Amputation rates and the reduction of lesions that do not heal will be assessed, with an evaluation of statistical significance between the groups. The evaluation of the domains of the SF-36 quality of life questionnaire will also be done in both groups. The data from this questionnaire upon admission and after three months of follow-up will be compared.
Data analysis
The collected data will be stored in a Microsoft Excel 2016 spreadsheet format. After checking for errors and inconsistencies, descriptive summaries will be provided by treatment arm, including absolute and relative frequencies and measures of central tendency and variability. We will do regular monitoring, with active search by address and phone to avoid sample loss. We will use the intention-to-treat in the analysis. Blinding will be done for the researcher who will evaluate the final data and for the statistician.
Logistic regression models will be used to assess associations between the categorical variables. Both relative effects (odds ratios) and absolute effects (risk differences) will be presented with corresponding confidence intervals. Student's t-test or a similar non-parametric method will be used for the analysis of continuous variables. All examinations will be performed at 5% significance in the IBM SPSS® program, Version 24.0, 2016 (IBM, Armonk, New York, USA).
Ethical aspects
This study will be based on the principles of Resolution 466/12 of the National Health Council that regulates the research involving human subjects. The patients involved shall be duly informed and clarified about the importance and purpose of the study; if patients accept to participate, they will sign an informed consent form (see supporting information). The non-participation in the study or waiver, privacy, reliability, and anonymity of the participants will be guaranteed.
The study was initiated after approval by the Research Ethics Committee of the ABC University. It is registered with the Brazilian Registry of Clinical Trials (ReBEC) under the number RBR-7bd3xy.
Adverse events
This study is being conducted in accordance with the guidelines of the Brazilian Society of Hyperbaric Medicine (SBMH) and Undersea & Hyperbaric Medical Society (UHMS), and if adverse events occur (such as dizziness, seizure, pneumothorax, pneumomediastinum, nauseas, middle ear barotrauma, seizure, ear pain, confinement anxiety, hypoglycemic event and shortness of breath), these will be conducted according to these guidelines, as well as be documented and published later.
{kind=link}