Soybean β-conglycinin α-subunit is an important allergen that adversely affects the nutritional and processing qualities of soya products. Although inheritance of the α-subunit and the molecular basis of α-null mutations have been studied intensively, the molecular mechanism that regulates α-subunit expression remains unclear. Here, we demonstrated that a long intergenic non-coding RNA, acting as a soybean cis-tether terminator1 (designated Linc-GmSTT1) regulate β-conglycinin α-subunit expression. The Linc-GmSTT1 was mapped in physical proximity of α-subunit CG-α-1 gene and demonstrated to be a crucial element of the convergent alpha-transcription termination unit (alpha-TTU). Ingeniously, by reading through, Linc-GmSTT1 and CG-α-1 gene co-transcribed and subsequently achieve its Cgy-2-locus (confirm α-normal) specific regulation function via Linc-GmSTT1-intermolcular interactome. This work provides a unique model whereby LincRNA regulated the effective transcriptional termination of proximal protein-coding genes which might be a crucial procession protecting it from the silencing machinery in plant.
Research Article
A cis-Tether Terminator, Linc-GmSTT1, Regulates Transcription Termination via the Linc-GmSTT1-intermolecular Interactome in Soybean
https://doi.org/10.21203/rs.3.rs-75237/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Linc-GmSTT1
cis-tether terminator
Transcription Termination-associated
Soybean β-conglycinin α-subunit
Beta-conglycinin is an abundant soybean seed-storage protein. The β-conglycinin subunit composition is important in determining the nutritional and processing qualities of soya products. The α-subunit of β-conglycinin adversely affects nutritional, processing, and allergenic qualities; therefore, soybean cultivars lacking this subunit are highly desirable to improve the nutritional and processing qualities of seed proteins1,2. The α-subunit is controlled by the Cgy-2 locus, which carries either Cgy-2 (α-normal; dominant) or cgy-2 (α-null; recessive) alleles3. The inheritance and molecular basis of α-null mutations at this locus have been studied intensively3–5. The α-subunit is encoded by CG-α-1 (Glyma.20g148300) and CG-α-2 (Glyma.20g148400), which are positioned in a tail-to-tail orientation in linkage group I/chromosome Gm20 (LGI/Chr20)4–6. The typical inverted repeated (IR) structure is named alpha-IR (AB604030), and a 3.3-kb intergenic region separates the two genes in the cultivar ‘Williams 82’5. Absence of the α-subunit is inherited independently, which can be incorporated into soybean cultivars and causes no physiological abnormalities3,7,8. Alpha-subunit gene expression is controlled at the transcriptional or post-transcriptional levels6,9. However, the molecular mechanism regulating α-subunit expression remains unclear.
Transcription comprises three stages (initiation, elongation, and termination) strongly controlled by numerous specific factors. Although initiation and elongation have been well studied10, termination remains poorly understood and the mechanism unclear, particularly for RNA polymerase (RNAP) II transcription. Termination is vital for release of RNAP from the template because it avoids interference with transcription of downstream genes11. Moreover, it ensures availability of a RNAPs pool for new transcription or re-initiation. Termination mechanisms range from relatively simple to extremely complex. The RNAPII-mediated termination is not effected at a conserved site or a constant distance from the 3′-end of mature RNAs. Termination can occur in mammals at any site from several to thousands of base pairs downstream from the mature RNA 3′-end12. RNAPII transcription termination occurs concurrently with pre-mRNA 3′-end processing13. In addition, an intact polyadenylation signal is essential for transcription termination of protein-coding genes in yeast and human cells14.
The mRNA 3′-end processing is vital for maturation of all mRNAs and is associated with splicing, transcription, and termination13,15. More than 14 proteins participate in the mammalian pre-RNA 3′-end processing machinery within a 1 MDa complex16,17. Among these proteins, CPSF (cleavage and polyadenylation-specific factor) is the AAUAAA signal recognition protein. Various factors link 3′-end processing to transcription, such as cyclin-dependent kinase (CDK) and the C-terminal domain (CTD) of the RNAPII largest subunit. The pre-mRNA is protected from exonuclease degradation by PABP (poly(A)-binding protein). The pre-mRNA is also needed for efficient and correct poly(A) tail synthesis. In addition, Clp1 interacts with CPSF complexes18 and CFI (cleavage factor I) and shows RNA kinase activity19. The CstF (cleavage stimulation factor) comprises three subunits (CstF-50, CstF-64, and CstF-77). The third subunit directly recognizes the U/GU-rich factor. Fip1 binds with CPSF-160, directly recognizing AAUAAA20. The Paf1 complex contributes to modification of chromatin associated with transcription21.
Long intergenic non-coding RNAs (lincRNAs) are a class of regulatory RNAs with diverse biological functions. Some lincRNAs contribute to gene silencing mechanisms, such as imprinting22 or dosage compensation23 and other events24. The co-transcription of non-coding transcripts (termed transcriptional interference) can affect adjacent gene transcription and has been reported primarily in yeast SER3 genes25. The prevalent interpretation is that the intrusive transcription procedure, not the transcript itself, affects transcription of the adjacent coding genes. However, a certain gene locus can be activated by the transcription products of the intergenic non-coding region, counteracting silencing by polycomb-group proteins26. An alternative hypothesis is that the equivalent chromatin is maintained by non-coding RNA transcription within a transcriptionally active state by preventing attachment of repressive protein complexes to the cis-regulatory element26,27.
Here, we describe a unique regulatory mechanism of normal β-conglycinin α-subunit expression by lincRNA-mediated intergenic non-coding region convergent transcription termination. By comparing genome-wide lincRNA expression profiles in soybean ‘DongNong47’ (DN47; α-normal) and its α-null near-isogenic line (NIL), we identified Linc-GmSTT1 as involved in regulating the absence or presence of the α-subunit. Ingeniously, by reading through, Linc-GmSTT1 and CG-α-1 gene co-transcribed and subsequently achieve its Cgy-2-locus (confirm α-normal) specific regulation function via Linc-GmSTT1-intermolcular interactome. This work provides a unique model whereby LincRNA regulated the effective transcriptional termination of proximal protein-coding genes which might be a crucial procession protecting it from the silencing machinery in plant.
Identification and characterization of Linc-GmSTT1. The α-subunit gene expression is controlled at the transcriptional and post-transcriptional levels6,9. To identify long non-coding RNAs (lncRNAs) associated with the α-null trait, we used RNA sequencing (RNA-Seq) to assess the genome-extensive lncRNA expression profiles in DN47 compared with its α-null NIL. The lincRNA transcript MSTRG128686 (designated Linc-GmSTT1) was located in the intergenic non-coding region within the α-subunit genes CG-α-1 and CG-α-2 in the Cgy-2 locus (AB604030) of DN47 (Fig. 1A, B). Linc-GmSTT1 was located 27 bp downstream of the 3′ untranslated region (UTR) of CG-α-1 in DN47 (Fig. 1A, B), but was not detected in NIL (Fig. 1D). Linc-GmSTT1 cis-regulated the differential expression of seven coding genes (Fig. 1A, Table S1). To obtain the full-length cDNA corresponding to MSTRG128686, we performed 5′ and 3′ RACE. The cDNA from developing seeds of DN47 (20 days after flowering; DAF) was amplified using MSTRG128686-specific primers and sequenced; the product (1212 bp) was designated Linc-GmSTT1 (Fig. S1). It contained 84 bp of the 5′ UTR, two exons, one intron, and 81 bp of the 3′ UTR (Fig. 1C). Quantitative real-time PCR (qRT-PCR) results agreed with the RNA-Seq data, showing that Linc-GmSTT1 expression was significantly lower in NIL than DN47 (Fig. 1E). Thus, the presence or absence of Linc-GmSTT1 was associated with the α-subunit normal or null phenotype.
Identification of Linc-GmSTT1 in DN47 led us to compare the intergenic non-coding region of the Cgy-2 locus between DN47 and NIL (Fig. 1B). In addition to the coding-region IR of the alpha-IR locus reported previously5, we detected two intergenic terminal inverted repeat (ITIR) units (Fig. 1B) that were identical in DN47 and NIL. The first unit, located immediately downstream of the 3′ UTR-end of the CG-α-1 flanking region, was a 27-bp ITIR1(L) fragment. The corresponding inverted repeat sequence of ITIR1(L), designated ITIR1(R), was embedded in the 3′ UTR of CG-α-2 (Fig. 1B, red arrowheads). The second ITIR was designated ITIR2(L)/ITIR2(R). ITIR2(L) was a 364-bp sequence located upstream of Linc-GmSTT1 and comprised 84 bp within the 5′ UTR region and 280 bp extending into exon 1 of Linc-GmSTT1, whereas ITIR2(R) was located in reverse orientation in proximity to the 3′ UTR-end of CG-α-2 (Fig. 1B, green arrowheads). These ITIR units might produce a double-stranded RNA molecule potentially involved in the silencing regulatory pathways.
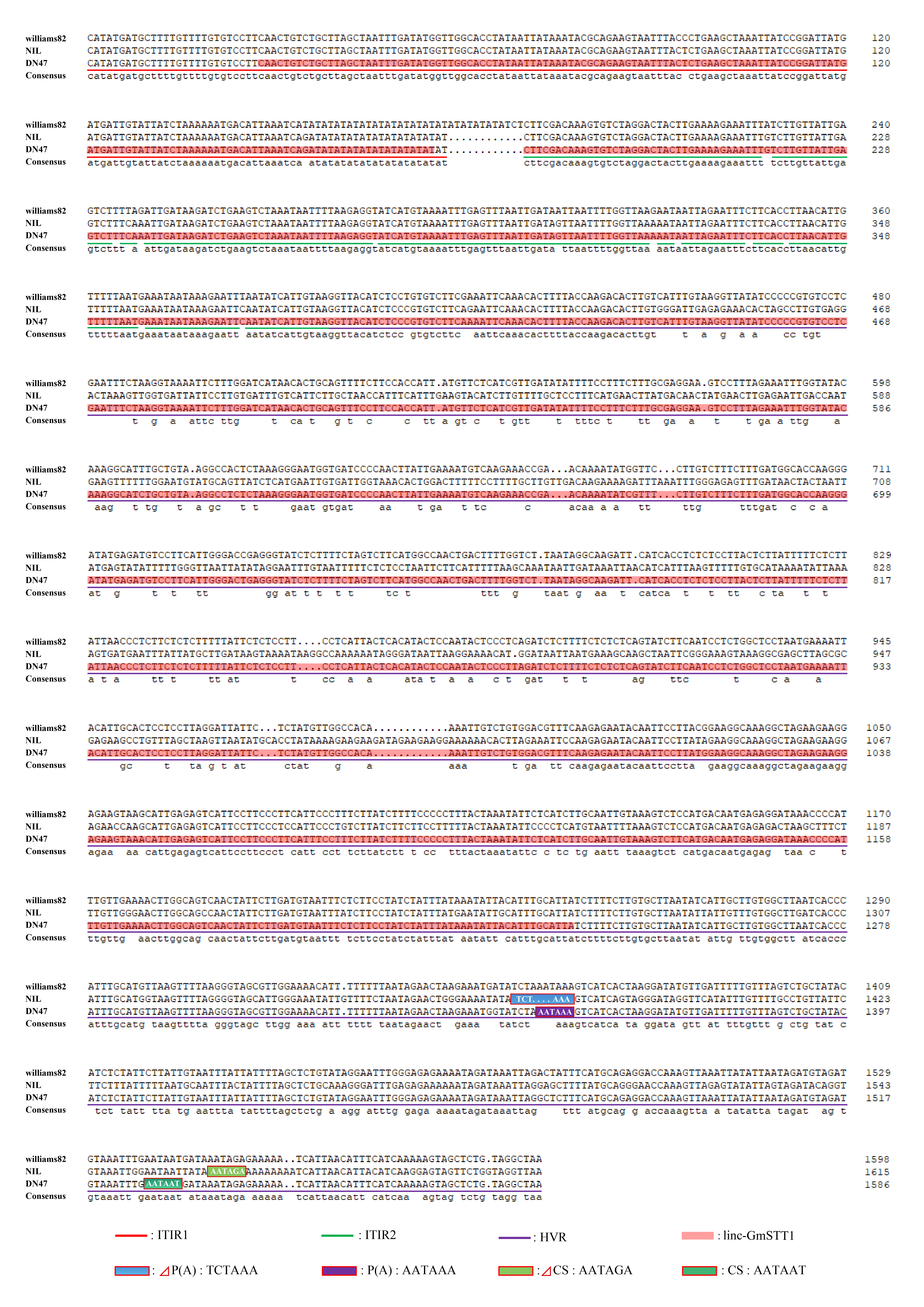
Although the coding sequences of the two α-subunit genes were identical in DN47 and NIL, we observed striking differences in the intergenic non-coding region from nt 38681395 to 38682596. We named this region HVR (hypervariable region; Fig. 1B). The HVR co-segregated with absence of the α-subunit. Furthermore, the first half of the HVR in NIL corresponded to the latter part of exon 1 of Linc-GmSTT1 (nt 38681395 to 38682244) in DN47. The HVR nucleotide sequence was 62.16% identical to the corresponding sequence in Linc-GmSTT1 (Fig. S2). In addition, two termination-related motif differences were observed: the poly(A) signal [P(A)] 5′-AATAAA-3′ (nt 38682359 to 38682364) and the cleavage site (CS) element 5′-ATTAAT-3′ (nt 38682512 to 38682517) located, respectively, 114 bp and 267 bp downstream of the 3′ UTR of Linc-GmSTT1 in DN47 were mutated in NIL to ΔP(A): 5′-TCTAAA-3′ and ΔCS: 5′-ATTAGT-3′, respectively (Fig. 1B, Fig. S2). The Linc-GmSTT1-deficient genotype and the mutations in both the functional P(A): AATAAA signal site and the CS: ATTAAT site co-segregated with the α-null phenotype, implying a close association with the molecular basis of the α-null trait. These data provided evidence for (i) existence of a novel natural convergent alpha-TTU (nt 38680997 to 38682596) that included ITIR1(L), ITIR2(L), and HVR, and (ii) Linc-GmSTT1 being the core of alpha-TTU (Fig. 1B, orange box).
Convergent transcription termination of CG-α-1 and Linc-GmSTT1 genes. The CG-α-1 gene was closely associated with lincRNA because the Linc-GmSTT1 gene was located only 27 bp from the 3′ UTR-end of CG-α-1 (Fig. 1A). Closely spaced genes are particularly prone to inducing occlusion and interference in transcription, especially when expressed simultaneously28. Normal expression of CG-α-1 and Linc-GmSTT1 in DN47 was indicated to be the result of a balance between two factors: (1) transcriptional interference and (2) initiation of transcription of the downstream Linc-GmSTT1. We speculated that co-transcription of Linc-GmSTT1 and CG-α-1 resulted in nascent transcripts from the adjacent sequences including CG-α-1 and Linc-GmSTT1, whereby the CG-α-1/Linc-GmSTT1 cryptic transcripts might be the first step required for initiation and termination of Linc-GmSTT1 transcription as well as regulating transcription termination of the Cgy-2 locus.
To assess the possibility of co-transcription, we searched for evidence of transcriptional readthrough of CG-α-1 into the Linc-GmSTT1 gene using RT-PCR amplification followed by sequencing. A CG-α-1-specific forward primer (F1) located within the 3′ UTR of CG-α-1 was designed; in addition, eight reverse primers were designed at five locations (R1–R5) within the alpha-TTU region, R6 and R7 within the intergenic region downstream of alpha-TTU, and R8 within the 3′ UTR of CG-α-2 (Fig. 2A, Table S2). The PCR products were amplified using the F0+R0 and F1+R1–R5 primer pairs, but not the F1+R6–R8 primer pairs (Fig. 2B). The amplified sequences were consistent with the target fragment sequences (Data S1). These data indicated transcription of CG-α-1 continued through the intergenic region and extended into Linc-GmSTT1. Therefore, we speculated that through sharing a common alpha-TTU with CG-α-1, Linc-GmSTT1 might effectively complete both initiation and termination, simultaneously contributing to regulation of transcription termination of the Cgy-2 locus.
CRISPR/Cas9-mediated targeted mutagenesis of Linc-GmSTT1. To determine whether mutation of Linc-GmSTT1 was responsible for the α-null mutant phenotype, we used CRISPR/Cas9 technology to knockout the Linc-GmSTT1 gene in the ‘DongNong 50’ (DN50) background (α-subunit normal) (Fig. 3). Genomic DNA was extracted from 25 independent transgenic T0 seeds, which were used to amplify the Linc-GmSTT1 gene region by PCR (Fig. 3B), followed by DNA sequencing to validate the targeted Linc-GmSTT1 gene disruption in the transgenic T0 plants. Nineteen T0 transgenic events contained a CRISPR/Cas9-edited large fragment deletion (Fig. 3C). One example (event 11, Linc-GmSTT1-13a) is illustrated in Fig. 3B, D–F. Event 11 represented a homozygous 551 bp deletion in the Linc-GmSTT1 gene (Fig. 3B, D).
We selected T1 seeds harboring CRISPR/Cas9-induced homozygous mutation of Linc-GmSTT1 for subunit-phenotypic analysis; the results for Linc-GmSTT1-13a are illustrated in Fig. 3E. Knockout of the major 551 bp fragment of Linc-GmSTT1 led to deletion of the β-conglycinin α-subunit in Linc-GmSTT1-13a T1 seeds (Fig. 3E, red box). Thus, mutation of Linc-GmSTT1 affected expression of the β-conglycinin α-subunit in seeds, suggesting that intact Linc-GmSTT1 is essential for expression of the α-subunit. The T1 seeds of Linc-GmSTT1-13a germinated well and showed no obvious growth abnormality (Fig. 3F).
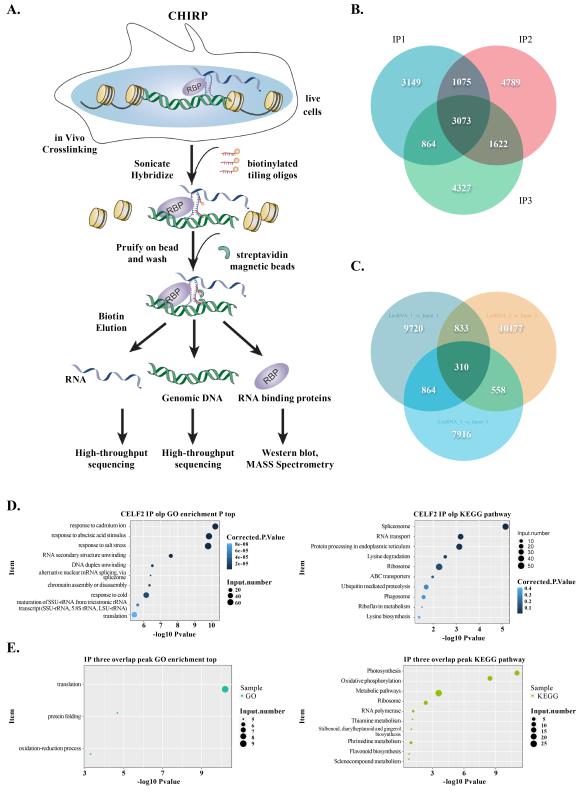
Interaction of RNAs, DNA, and proteins with Linc-GmSTT1. Chromatin isolation by RNA Purification (ChIRP) is a scalable new technique to dissect pairwise RNA–DNA, RNA–RNA and RNA–protein interactions in living cells (Fig. S3A). For further comprehension of the potential mechanism included in the regulation of Linc-GmSTT1, we used the ChIRP-DNA-Seq, ChIRP-RNA-Seq and ChIRP-MS analysis to investigate the intermolecular interactions Linc-GmSTT1-DNA, Linc-GmSTT1-RNA and Linc-GmSTT1-protein in developing soybean seeds (20 d after flowering) (Fig. S3A). In total, 3073 Linc-GmSTT1-interacting RNA peaks (Fig. S3B), 310 Linc-GmSTT1-interacting DNA peaks (Fig. S3C) and eight Linc-GmSTT1-interacting-proteins (Fig. 4A) were identified in three biological replicates.
Identification of Linc-GmSTT1–RNA and Linc-GmSTT1–protein interactions. Using ChIRP-Seq, we detected 3073 Linc-GmSTT1–RNA-interacting peaks (Fig. S3B), annotated as 2372 genes (Dataset S2).Go and KEGG analysis show that the most abundant Linc-GmSTT1–RNA interacting genes (LRGs) were involved in biological process response to cadmium ion (60 genes, Fig. S3D Left) and the majority of LRGs enriched in the spliceosome pathway (46 genes, Fig. S3D Right). These data indicate that Linc-GmSTT1 use numerous RNA–RNA intermolecular interactions to achieve its regulation function.
Using a genome-wide ChIRP–MS approach, we tested the interaction between Linc-GmSTT1 and endogenous proteins. A Venn diagram revealed that eight Linc-GmSTT1-interacting proteins were detected (Fig. 4A). Six were known proteins (I1LN32, I1LWR4, K7KHM4, I1JKI6, A0A0R0HL94, and A0A0R0JEA7) and two were non-characterized proteins (A0A0R0I621 and A0A0R0ILB2; Fig. 4A). Taking A0A0R0I621 (Glyma.09G092600) as an example, we verified the RNA–protein interaction detected by ChIRP-MS (Fig. 4B, C). Interaction between Linc-GmSTT1 and A0A0R0I621 was confirmed by RNA pull-down: the A0A0R0I621 protein was co-precipitated by an intro-transcribed biotinylated Linc-GmSTT1 RNA sense transcript (Fig. 4D). A yeast two-hybrid assay revealed that A0A0R0I621 interacted with I1JQR6 (Glyma.03G219200), I1LN46 (Glyma.11G253100), and I1LY15 (Glyma.13G106300; Fig. 4E) annotated with DNA replication licensing factor MCM7, DNA topoisomerase 3-beta, and a hypothetical protein, respectively.
Linc-GmSTT1 is a Cgy-2-allele-specific tether. We detected 310 Linc-GmSTT1–DNA-interacting peaks (Fig. S3C), annotated as 131 genes (Data. S3). The Linc-GmSTT1–DNA interacting genes (LDGs) were predominantly classified to the translation process categories within the biological process GO class (Fig. S3E-left). KEGG analysis revealed that some LDGs were involved in the ribosome and RNA polymerase pathway associated with transcriptional termination (Fig. S3E-right).
Intriguingly,We found Linc-GmSTT1 specifically bound to its own genomic sequence in the Cgy-2 locus in DN47 but not in NIL (Fig. 5A). Furthermore, we identified three Matrix motifs located within the genomic sequence of Linc-GmSTT1. The first motif contained GTTGG, the second contained ATAATTG, and the third contained TACAGT (Fig. 5B). Comparing the sequences with those in NIL, we found that these motifs were the novel putative Linc-GmSTT1-specific end-terminating cis-regulatory Matrix motifs that were detected in the cultivars Williams 82 and DN47, but not in the corresponding HVR region in NIL (Fig. 5B,C).
Emerging data indicate that lincRNAs can function as tethers. In theory, lincRNA has an intrinsic cis-regulatory capacity because it is able to function while tethered to its own locus29; by remaining tethered to the site of transcription, it can uniquely direct allelic regulation30-32. Our data indicate that α-subunit-associated Linc-GmSTT1 may recognize targets by DNA–RNA recognition, and Linc-GmSTT1 plays its role by binding to its own genomic sequence to function as a tether for the Cgy-2-locus-specific and allelic control (Fig. 6B). The discovery that Linc-GmSTT1 is a cis-tether terminator involved in transcription termination was unexpected because very few spontaneous lincRNA terminators of transcription termination have been described in plants.
Based on our research results in the current study, it is assumed that Linc-GmSTT1 is a key transcription terminator in the expression of α-subunit of β-conglycinin, and that genes involved in the transcription termination machinery might be potential targets of Linc-GmSTT1. Combining information from our ChIRP analysis databases and iTrack data, we identified 14 Linc-GmSTT1-interacting-DNA genes (LIDGs) that were annotated as CPSF (5 genes), CTD (3 genes), CDK (3 genes), PABP (2 genes) and Clp1 (1 gene). In addition, we found 45 Linc-GmSTT1-interacting-RNA genes (LIRGs) that were annotated as CPSF (13 genes), CTD (7 genes), CDK (11 genes), PABP (6 genes), Clp1 (cleavage factor I) (2 genes), CstF (1 gene), Fip1 (2 genes), and Paf (3 genes) (Table S3). Among these 59 genes associated with 3′-end processing machinery, the proteins encoded by 20 genes were confirmed to be differentially expressed between NIL and DN47 by proteomic differences in the iTrack comparative analysis (Table S3).
Recently, a study using full-length cDNA datasets from humans and mouse proved that lincRNAs predominantly originate from the vicinity of protein-coding genes, and transcription of certain lincRNAs depends on the same promoter regions as the nearby protein-coding genes11,28. Being positioned close to their target protein-coding genes, lincRNAs might depend on the same promoter regions to regulate expression of the protein-coding genes, which might be a common lincRNA-mediated regulatory mechanism in higher eukaryotes. In the present study, we demonstrated that Linc-GmSTT1 depend on the same transcriptional termination region with CG-α-1. Transcription of CG-α-1 continued through the intergenic region and the entire Linc-GmSTT1, resulting in nascent transcripts from adjacent sequences that included both CG-α-1 and Linc-GmSTT1 (Fig. 2). Moreover, the majority of 2372 LRGs were classified as spliceosome pathway genes (Fig. S3D-right). We speculate that chimeric CG-α-1/Linc-GmSTT1 cryptic transcripts first require precision splicing to release mature mRNAs of CG-α-1 and Linc-GmSTT1, which might explain the predominant classification of LRGs as spliceosome pathway genes. How the splicing of a single co-transcription unit leads to the Linc-GmSTT1 transcript distinct from that of the protein-coding gene CG-α-1 needs further research.
LincRNAs interact with numerous DNAs, RNAs and proteins for accurate transcriptional regulation33-42. Our data indicate that Linc-GmSTT1 has the unique capacity for interacting simultaneously with multiple DNA, RNA and protein molecules (Fig. 6B, C, D), and suggest that Linc-GmSTT1 acts at nearly every level of transcriptional regulation. These results provide strong evidence there is no single mechanism by which the α-subunit is strictly and effectively transcribed. However, we have not yet proven how the functions of Linc-GmSTT1 are coordinated cooperatively to interact with specific interactome including DNA, RNA and protein sequences and so regulate the expression of the α-subunit at different transcriptional levels. This issue will require further study.
The LincRNA-mediated intergenic region convergent transcription termination is a new model (Fig.6) for regulating the expression of soybean seed storage protein subunit. Based on the data presented here, we propose that a tail-to-tail α-subunit genes convergent terminator system operates at two levels of regulating the expression of the α-subunit: (1) co-transcription of CG-α-1 and Linc-GmSTT1 at transcriptional level (Fig.6A), and (2) post-transcriptional regulation of the transcription of the Cgy-2-locus specifically regulated by Linc-GmSTT1 (Fig. 6B, C, D) is a prerequisite for the normal expression of the α-subunit (Fig.6E).Both mutation (Fig.6F)and knockout (Fig.6G) of Linc-GmSTT1 results inefficient termination of α-subunit CG-α-1 gene (Fig.6H) and induces post-transcriptional α-subunit gene silencing (Fig.6I). The proper transcription termination of α-subunit might be a crucial process in protecting α-subunit gene from the silencing machinery. The possibility of intergenic lincRNA-mediated regulation in other similar tail-to-tail gene pairs is yet to be examined.
Acknowledgements
This research was supported by the Ministry of Science and Technology of China (grant no. 2016YFD0100500), China National Novel Transgenic Organisms Breeding Project (2016ZX08004-004-006), National Natural Science Foundation of China (31801386, 31371650, and 31071440), and funds from 2016RQYXJ018, 2017RAQXJ104, LC2018008, and 2018M641839. We would like to thank Jun Fang for critical reading of the manuscript and advice.
Author contributions
Conceptualization and experimental design: SS.L.; experimentation: B.S., TT.L., Z.P., M.Z., X.F. and B.N.; plant management and seed collection: JJ.W. and YH.Z.; formal data analysis: PF.X. and B.S.; writing–original draft: SS.L., B.S. and PF.X.; writing–review and editing: SS.L.; visualization: B.S.; supervision: SZ.Z.; funding acquisition: SZ.Z.
Competing interests
All authors declare they have no conflict of interest.
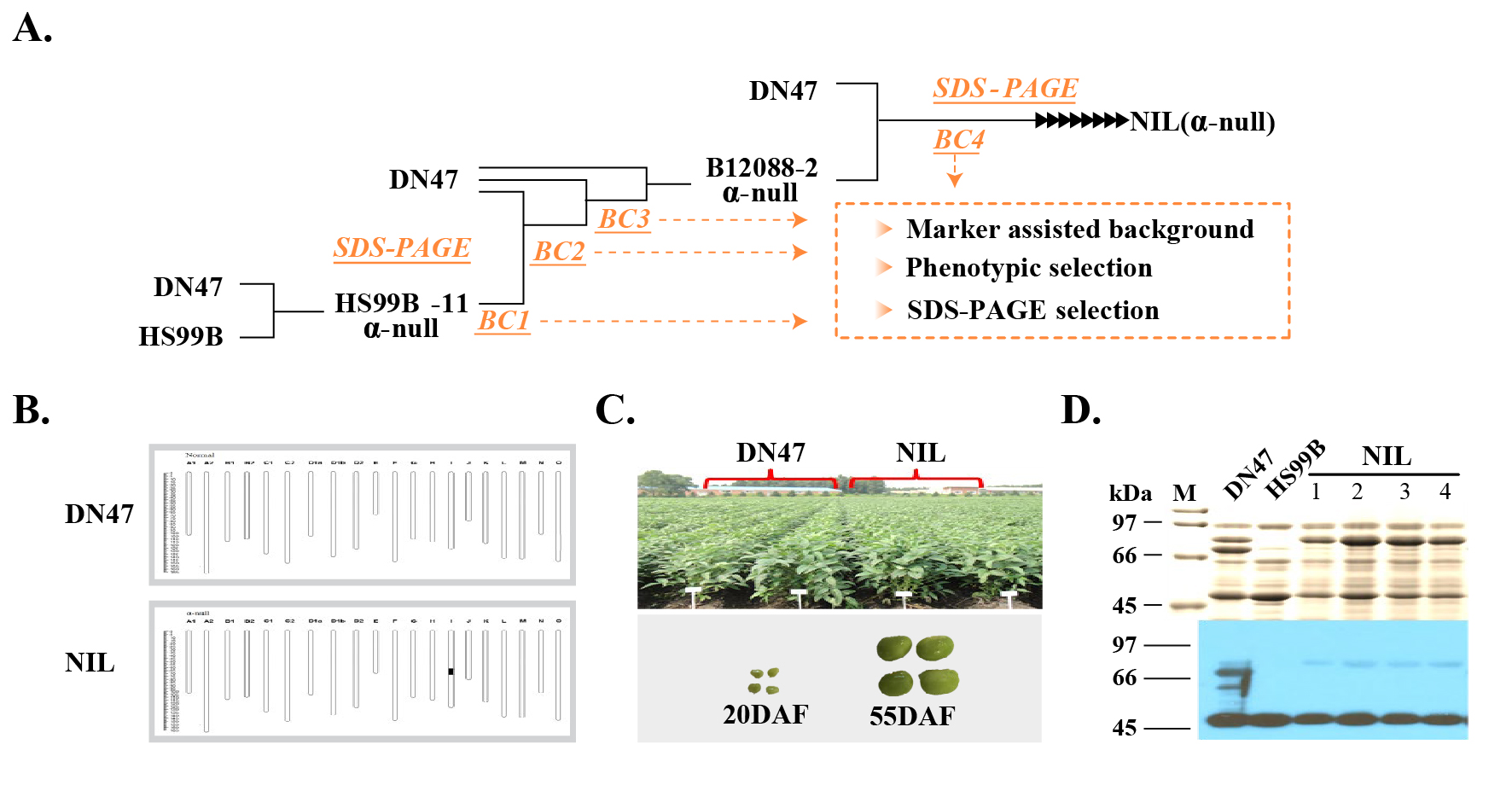
Plant material and growth conditions. The α-null type NIL used in this study was developed by four generations of backcrossing a line harboring cgy-2 (confirmed α-null) from RiB with DN47, followed by five generations of selfing to generate a BC4F5 NIL population (Fig. S4). We previously used this population to investigate α-null-related transcription-level changes8. Standard farming practices were used to grow the BC4F5 NIL plants in a randomized block design at the Northeast Agricultural University Experimental Station, China. Pod samples were collected during the seed development stage at 20 DAF (Fig. S4C) during the summer of 2018. SDS-PAGE and western blot analyses confirmed that the α-null phenotype was stably inherited in NIL (Fig. S4D). The BC4F5 seeds harvested in 2018 were used for the ChIRP analyses. ‘DongNong 50’ (DN50), a soybean cultivar that shows high transformation efficiency, was used for CRISPR/Cas9 analysis.
Phenotype screening for the α-subunit-null mutation in the NIL using SDS-PAGE analysis. The absence of the α-unit of b-conglycinin was confirmed in the collected NIL seed samples by analyzing the subunit composition of seed proteins by SDS-PAGE (Supplementary Fig. 1D). SDS sample buffer was used to extract seed proteins from a small amount of cotyledon tissue (5% [v/v] 2-mercaptoethanol, 2% [w/v] SDS, 5 M urea, 62.5 mM Tris amino methane, and 10% [w/w] glycerol). Samples were centrifuged at 15,000 ×g, after which 10 μL supernatant was used in 12.5% [w/w] separating and 4.5% [w/w] stacking polyacrylamide gels that were stained using Coomassie Brilliant Blue R 250.
RNA quality testing. Developing seeds harvested from DN47 and NIL plants at 20 days after flowering during the summer of 2018 were used for RNA-Seq. Total RNA was extracted using an enhanced cetyltrimethylammonium bromide (CTAB) method. We checked RNA quality using a K5500® spectrophotometer (Kaiao, Beijing, China). The RNA integrity was assessed and RNA concentrations were calculated using an RNA Nano 6000 Assay Kit for a Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA).
Library preparation for lincRNA sequencing. A sample (3 μg) of extracted RNA was used as the initial material. Epicentre Ribo-Zero™ Gold Kits (Human/Mouse/Rat/Other) (Epicentre, Madison, WI, USA) were used to remove ribosomal RNA. Sequencing libraries (with different index labels) were subsequently created using a NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA).
Library checking. A Qubit® RNA Assay Kit was used to measure the RNA concentrations of the prepared libraries, after which samples were diluted to 1 ng/μL. Using an Agilent Bioanalyzer 2100 system (Agilent Technologies), the insert sizes were evaluated, and appropriate inserts were quantified using a TaqMan fluorescence probe and a StepOne Plus Real-Time PCR System (Applied Biosystems) (valid library concentration > 10 nM).
Library clustering and sequencing. A cBot cluster-generation system with a TruSeq PE Cluster Kit (version 4) cBot-HS (Illumina, San Diego, CA, USA) was used to complete the clustering of the index-coded samples. The libraries were sequenced on an Illumina platform after clustering to generate 150-bp paired-end reads.
Data quality control. Perl scripts were used to process the raw data to guarantee the suitable quality of the data for subsequent analyses. The reference genome and the annotation files were downloaded from the ENSEMBL database (http://www.ensembl.org/index.html). The genome index was used to build Bowtie2 (version 2.2.3). Using TopHat (version 2.0.12), clean sequence data were mapped to the reference genome. The latter program was also used to recognize exon–exon junctions by separating the mapped reads and remapping them to the reference genome. TopHat uses Bowtie2 for mapping, which improves the accuracy and speed of the analysis.
Quantification of gene expression levels. Read counts for each gene in every sample were determined using HTSeq (version 0.6.0), after which the number of reads per kilobase per million mapped reads (RPKM) was computed with the following equation to approximate the gene expression levels in each sample:

where R represents the number of reads for a particular gene in a specific sample, N denotes the total number of mapped reads in a specific sample, and L is the length of a particular gene.
Analysis of differentially expressed genes. The DESeq (version 1.16) program was used to analyze DEGs in DN47 and NIL in accordance with a negative binomial distribution model. A P-value was allocated to each gene and the Benjamini–Hochberg method used to control the false discovery rate. Genes with |log2 ratio| ≥ 1 and q ≤ 0.05 were recognized as DEGs.
Quantitative real-time PCR validation. Total RNA was transcribed reversely into cDNA utilizing SuperScript III Reverse Transcriptase (Invitrogen, Grand Island, NY, USA) following the manufacturer’s instructions A 2× PCR Master Mix and Applied Biosystems ViiA 7 Real-Time PCR System were used for qRT-PCR analysis with incubation for 10 min at 95°C, followed by 40 cycles of 60°C for 1 min and 95°C for 10 s. The 2−ΔΔCt method was used to calculate the relative mRNA and lincRNA expression levels, which were normalized to GAPDH as an endogenous reference transcript. The data shown represent the means of three repetitions.
5′ and 3′ RACE of MSTRG128686. The 5′ RACE PCR amplification was performed based on the Invitrogen 5′ RACE system manual. For cDNA first-strand synthesis, the mixture contained 5 μL total RNA and incubation was performed for 1 μL random primer at 70°C for 5 min followed by placement in an ice bath for 2 min. Then, 2.0 μL of 5× first-strand buffer, 0.5 μL of 10 mM dNTPs, 0.25 μL RNase inhibitor, and 0.25 μL reverse transcriptase were added. The mixture was made to 10.0 μL total volume and incubated at 42°C for 60 min followed by 72°C for 10 min. For 5′ RACE with a nested PCR reaction system (end C method), reverse transcription used specific primers RC583-RT1/RC583-RT2 to amplify the cDNA, and after the RNase H and TdT treatment we performed nested PCR (see the following section). For 5′ and 3′ RACE of rare cDNAs, the temperature parameters for PCR were: 3 min at 95°C followed by 33 cycles at 94°C for 30 s and 68°C for 30 s; after a 7-min ultimate extension at 72°C, the PCR was repeated.
The 3′ RACE amplification was also conducted using nested PCR, using the 3′ adaptor as the reverse primer, cDNA as the template, and the same conditions and cycle parameters as for 5′ RACE, except that the annealing temperature was 58°C for 30 s. The PCR products were separated on 1.0% (w/w) agarose/ethidium bromide gels in 1× TBE buffer containing 90 mM Tris-borate and 2 mM EDTA (pH 8.0 at 22°C). We used a 1 kb DNA ladder as a DNA size marker.
RT-PCR. Total RNA was extracted from DN47 and NIL seeds at 20 days after flowering using TRIzol reagent (Invitrogen) followed by treatment with RNase-free DNase I (Invitrogen) to eliminate genomic DNA. Treated RNA was utilized for RT-PCR. The RT-PCR amplification of the convergent transcription readthrough of CG-α-1/Linc-GmSTT1 transcripts was conducted using the primer pairs listed in Supplementary Table 2. The PCR-created products were cloned directly into pCRII using a TOPO TA cloning kit (Invitrogen) and subsequently sequenced.
CRISPR/Cas9-mediated Linc-GmSTT1 knockout. CRISPR/Cas9 gene-knockout constructs were developed using the pCBSG015(Basta) vector. We designed two sgRNAs targeting Linc-GmSTT1 at two locations: 5′-CTTACAAATGACAAGTGTCTTGG-3′ and 5′-GTTGGCCACAAAATTGTCTGTGG-3′. The two sgRNAs were added using pCBSG015(Basta) containing Cas9. The constructs were individually transformed into the DN50 (α-normal) background using soybean embryo cotyledonary node transformation.
The Cas9/sgRNA expression vectors in pCBSG015(Basta) were introduced into Agrobacterium tumefaciens strain EHA105 by electroporation. Embryo cotyledonary nodes from DN50 seeds germinated for 5 days were placed in a petri dish containing 50 mL Agrobacterium suspension. About 150 explants were treated for 2 h, and were then left at room temperature for 30–60 min for infection. After infection, the Agrobacterium liquid was discarded, the explants were transferred to the co-cultivation medium and incubated in the dark at 23°C for 3 days. After co-cultivation, the embryos were transmitted to the shoot-induction medium, cultured at 25°C for 7 days, then placed on selection medium containing glufosinate. After culture for 3 weeks, the glufosinate-resistant shoots were transferred to shoot-elongation medium containing glufosinate and cultured in the light for 6–9 weeks. The regenerated elongated seedlings were transferred to rooting medium at 25°C and cultured under light (5000 lux) until rooting.
For each transformed plant, to validate the CRISPR/Cas9-mediated gene disruption, genomic DNA was extracted from the leaves using the CTAB method. The target Linc-GmSTT1 gene fragment was amplified by PCR using the primer pair 5′-CTTCAACTGTCTGCTTAGCTAATTT-3′ and 5′-CCTTTGCCTTCCATAAGGAATTGT-3′. Ultimately, the PCR products were sequenced to verify the successful editing of the gene. Only transformed plants in which the target gene was edited successfully were used in the subsequent tests.
Crosslinking and chromatin preparation. One gram of frozen tissue was sliced and resuspended in 1 volume PBS, crosslinked in 1% (v/v) formaldehyde for 10 min, then quenched for 5 min with 0.125 M glycine, and collected by centrifugation at 2000 ×g for 5 min. Nuclei were lysed (100 mg/mL in nuclear lysis buffer: 50 mM Tris [pH 7.0], 1% [w/v] SDS, 10 mM EDTA, with DTT and PMSF added just before use) on ice for 10 min, and sonicated utilizing a Bioruptor until most chromatin was solubilized and the DNA was within the size range of 100–500 bp. Chromatin preparations were snap-frozen in liquid nitrogen and stored at −80°C until use.
Hybridization and washing. Chromatin was diluted in two volumes of hybridization buffer (1% [w/v] SDS, 750 mM NaCl, 1 mM EDTA, 15% [v/v] formamide, 50 mM Tris [pH 7.0], with DTT and PMSF added just before use). Probes (100 pmol) were added to 3 mL diluted chromatin and combined by end-to-end shaking at 37°C for 4 h. Streptavidin–magnetic C1 beads were rinsed three times in nuclear lysis buffer, then 100 µL of washed beads was added per 100 pmol probes, and the blend was mixed at 37°C for 1 h. Beads:biotin-probes:RNA:chromatin adducts were captured using magnets (Invitrogen) and rinsed five times with 1 mL wash buffer (0.5% [w/v] SDS, 2× SSC, with DTT and PMSF added just before use). At the last wash, the beads were resuspended. Aliquots of 300 μL were removed for isolation of protein, RNA, and DNA. All tubes were placed on a DynaMag-2 magnetic strip and the wash buffer was removed. After brief centrifugation, tubes were placed on a magnet strip and the last remnants of wash buffer were removed using a fine 10 μL pipette tip.
ChIRP protein elution and MS analysis. Beads were resuspended in 3× original volume of DNase buffer (0.1% NP-40 and 100 mM NaCl). Protein was eluted with 0.1 U/µL RNase H (Epicenter), 100 U/mL DNase I (Invitrogen), and a cocktail of 100 µg/mL RNase A (Sigma-Aldrich) at 37°C for 30 min. Protein eluent was supplemented with 0.2 volume of 5× SDS loading buffer, boiled for 5 min, separated on a NuPAGE 4%–12% (w/w) Bis-Tris gel, followed by silver staining to identify differential bands. The whole gel lane was excised, trypsinized, reduced, alkylated, and further trypsinized at 37ºC overnight. The resulting peptides were extracted, concentrated, and HPLC-purified. The peptides separated by liquid-phase chromatography were ionized through a nanoESI source and then passed through a tandem mass spectrometer LTQ Orbitrap Velos (Thermo Fisher Scientific, San Jose, CA, USA) with data-dependent acquisition- (DDA-) mode detection. Protein identification aligned the experimental MS/MS data with the theoretical MS/MS data from a database. Raw MS data were converted into a peak list and then used to search for matches in the database with strict filtering and quality control to produce possible protein identifications. The final protein identification list was used for functional annotation analysis using the GO and KEGG databases.
ChIRP DNA elution and high-throughput sequencing. Beads were resuspended in 3× original volume of DNA elution buffer (1% [w/v] SDS, 50 mM NaHCO3, and 200 mM NaCl), including DNA INPUT, and DNA was eluted with 100 µg/mL RNase A (Sigma-Aldrich) and 0.1 unit/µL RNase H (Epicenter). Elution was performed two times [for 1 h] at 37°C with end-to-end shaking, and both eluates were combined. Chromatin was reverse-crosslinked with formaldehyde at 65°C overnight then treated with 0.2 U/µL of proteinase K at 55°C for 60 min. DNA was then extracted with an equivalent volume of phenol:chloroform:isoamyl alcohol (Invitrogen) and precipitated with ethanol at −80°C overnight. Using a DNA library preparation protocol, eluted DNA was amplified into sequencing libraries based on the manufacturer’s instructions (KAPA). To create 151 nt paired-end reads, the recovered libraries were sequenced on an Illumina NextSeq 500 platform (ABLife Inc., Wuhan, China). The raw reads were ranged by Bowtie2 (version 2.2.9) with the Glycine max reference genome. The exclusively mapped reads were exposed to the peak-calling algorithm MACS (version 1.4.2) with default factors.
ChIRP RNA elution and high-throughput sequencing. Beads were resuspended in 95 μL RNA PK buffer (10 mM Tris-Cl [pH 7.0], 100 mM NaCl, 0.5% [w/v] SDS, and 1 mM EDTA), then 5 μL of proteinase K was added and the mixture was incubated at 50°C for 45 min with end-to-end shaking. For RNA INPUT samples (10 μL), 85 μL RNA PK buffer was added. All tubes were centrifuged briefly and heated at 95°C for 10 min, and then RNA was extracted with TRIzol:chloroform. Eluted RNA was amplified into sequencing libraries via a RNA library preparation protocol based on the manufacturer’s instructions (KAPA). To create 151 nt paired-end reads (ABLife Inc., Wuhan, China), the recovered libraries were sequenced on an Illumina NextSeq 500 platform. The raw reads were aligned by Bowtie2 (version 2.2.9) with the Glycine max reference genome. The exclusively mapped reads were exposed to the peak-calling algorithm MACS (version 1.4.2) with default factors.
- Ogawa, T., Bando, N., Tsuji H., Nishikawa, K. & Kitamura, K. α-Subunit of β-conglycinin, an allergenic protein recognized by IgE antibodies of soybean-sensitive patients with atopic dermatitis. Biosci. Biotechnol. Biochem. 59, 831-833 (1995).
- Krishnan, H. B., Kim, W. S., Jang, S. & Kerley, M. S. All three subunits of soybean β-conglycinin are potential food allergens. J. Agric. Food Chem. 57, 938-943 (2009).
- Takahashi, K., Mizuno, Y., Yumoto, S., Kitamura, K. & Nakamura, S. Inheritance of the α-subunit deficiency of β-conglycinin in soybean (Glycine max L. MERRILL) line induced by γ-ray irradiation. Jpn. J. Breed. 46, 251-255 (1996).
- Yoshino, M. et al. Structural variation around the gene encoding the α subunit of soybean β-conglycinin and correlation with the expression of the α subunit. Breed. Sci. 52, 285-292 (2002).
- Tsubokura, Y. et al. The β-conglycinin deficiency in wild soybean is associated with the tail-to-tail inverted repeat of the α-subunit genes. Plant Mol. Biol. 78, 301-309 (2012).
- Harada, J. J., Barker, S. J. & Goldberg, R. B. Soybean β-conglycinin genes are clustered in severa1 DNA regions and are regulated by transcriptional and posttranscriptional processes. Plant Cell 1, 415-425 (1989).
- Song, B. et al. Marker-assisted backcrossing of a null allele of the α-subunit of soybean (Glycine max) β-conglycinin with a Chinese soybean cultivar (a). The development of improved lines. Plant Breed. 133, 638-648 (2014).
- Song, B. et al. Transcriptome profile of near-isogenic soybean lines for β-conglycinin α-subunit deficiency during seed maturation. PLoS ONE 11, e0159723 (2016).
- Teraishi, M. et al. Suppression of soybean β-conglycinin genes by a dominant gene, Scg-1. Theor. Appl. Genet. 103, 1266-1272 (2001).
- Sims, R. J. III., Belotserkovskaya, R. & Reinberg, D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 18, 2437-2468 (2004).
- Greger, I. H., Aranda, A. & Proudfoot, N. Balancing transcriptional interference and initiation on the GAL7 promoter of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 97, 8415-8420 (2000).
- Proudfoot, N. J. How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem. Sci. 14, 105-110 (1989).
- Buratowski, S. Connections between mRNA 3′ end processing and transcription termination. Curr. Opin. Cell Biol. 17, 257-261 (2005).
- Connelly, S. & Manley, J. L. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev. 2, 440-452 (1988).
- Proudfoot, N. J., Furger, A. & Dye, M. J. Integrating mRNA processing with transcription. Cell 108, 501-512 (2002).
- Mandel, C. R., Bai, Y. & Tong, L. Protein factors in pre-mRNA 3′-end processing. Cell Mol. Life Sci. 65, 1099-1122 (2008).
- Shi, Y. et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 33, 365-376 (2009).
- De Vries, H. et al. Human pre-mRNA cleavage factor IIm contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 19, 5895-5904 (2000).
- Weitzer, S. & Martinez, J. hClp1: a novel kinase revitalizes RNA metabolism. Cell Cycle 6, 2133-2137 (2007).
- Kaufmann, I., Martin, G., Friedlein, A., Langen, H. & Keller, W. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 23, 616-626 (2004).
- Mueller, C. L., Porter, S. E., Hoffman, M. G. & Jaehning, J. A. The Paf1 complex has functions independent of actively transcribing RNA polymerase II. Mol. Cell 14, 447-456 (2004).
- Pauler, F. M. & Barlow, D. P. Imprinting mechanisms—it only takes two. Genes Dev. 20, 1203-1206 (2006).
- Heard, E. & Disteche, C. M. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 20, 1848-1867 (2006).
- Zaratiegui, M., Irvine, D. V. & Martienssen, R. A. Noncoding RNAs and gene silencing. Cell 128, 763-776 (2007).
- Martens, J. A., Laprade, L. & Winston, F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429, 571-574 (2004).
- Schmitt, S., Prestel, M. & Paro, R. Intergenic transcription through a Polycomb group response element counteracts silencing. Genes Dev. 19, 697-708 (2005).
- Sessa, L. et al. Noncoding RNA synthesis and loss of Polycomb group repression accompanies the colinear activation of the human HOXA cluster. RNA 13, 223-239 (2007).
- Khachane, A. N. & Harrison P. M. Mining mammalian transcript data for functional long non-coding RNAs. PLoS ONE 5, e10316 (2010).
- Lee, J. T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 23, 1831-1842 (2009).
- Jeon, Y. & Lee, J. T. YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146, 119-133 (2011).
- Yu, Y. et al. Panoramix enforces piRNA-dependent cotranscriptional silencing. Science 350, 339-342 (2015).
- Quinodoz, S. & Guttman, M. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol. 24, 651-663 (2014).
- Carrieri, C. et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491, 454-457 (2012).
- Yap, K. L. et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by Polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662-674 (2010).
- 35 Bierhoff, H., Schmitz, K., Maass, F., Ye, J. & Grummt, I. Noncoding transcripts in sense and antisense orientation regulate the epigenetic state of ribosomal RNA genes. Cold Spring Harb. Symp. Quant. Biol. 75, 357-364 (2010).
- Beckedorff, F. C. et al. The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet. 9, e1003705 (2013).
- Mercer, T. R. & Mattick, J. S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 20, 300-307 (2013).
- Pasmant, E., Sabbagh, A., Vidaud, M. & Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 25, 444-448 (2011).
- Poliseno, L. et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033-1038 (2010).
- Liu, T. et al. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1-AS expression. Mol. Med. Rep. 10, 1275-1281 (2014).
- Ray, D. et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172-177 (2013).
- Park, J. Y. et al. Roles of long non-coding RNAs on tumorigenesis and glioma development. Brain Tumor Res. Treat. 2, 1-6 (2014).
- Table.S1..docx
Seven differentially expressed genes were cis-regulated by Linc-GmSTT1 as identified by RNA-Seq
- Table.S2.docx
Primer sets used for RT-PCR amplification to prove transcription read-through of CG-α-1/Linc-GmSTT1
- Table.S3..docx
Endogenous Linc-GmSTT1 target genes involved in the transcription termination 3′-end processing machinery as identified by ChIRP analysis
- DatasetS1.docx
To confirm the co-transcribe between CG-α-1 and Linc-GmSTT1 gene, the amplified bands were cloned and sequenced, and the sequencing results were consistent with the target fragment sequences
- datasetS2..xlsx
Gene annotation of 2372 Linc-GmSTT1 ChIRP–RNA overlaps.
- datasetS3.xlsx
Gene annotation of 131 Linc-GmSTT1 ChIRP–DNA overlaps.
- Fig.S1.jpg
Figure S1 Nucleotide sequence of the Linc-GmSTT1 gene. Linc-GmSTT1 was located in the Chr20+ 38681025–38682244 region. Yellow-marked sequences indicate 5′ - and 3′ - non-translated regions. Non-marked sequences are Linc-GmSTT1 exon1 and exon2. Green marked sequence indicates Linc-GmSTT1 spliced intron.
- FigS2.jpg
Fig. S2 Alignment of alpha-TTU nucleotide sequences of the genotypes Williams 82, NIL, and DongNong 47 (DN47). The red underline represents the left portion of intergenic terminal inverted repeat 1 [ITIR1(L)], the green underline indicates the left portion of intergenic terminal inverted repeat 2 [ITIR2(L)], and the purple underline marks the hypervariable region (HVR). The HVR region associated with the α-null phenotype co-segregated with absence of the α-subunit of β-conglycinin. The HVR (nt 38681395 to 38682596) in NIL corresponded to downstream of ITIR2(L) in exon 1 of Linc-GmSTT1 in DN47; the HVR nucleotide sequence was 62.16% identical to that of the corresponding sequence in Linc-GmSTT1. Red shading in the DN47 sequence indicates Linc-GmSTT1, purple shading in the DN47 sequence indicates the sequence of the Poly Adenylation signal [P(A)]:AATAAA, blue shading in the NIL sequence marks the ΔP(A) sequence TCTAAA, dark green shading in the DN47 sequence represents the cleavage site (CS): AATAAT, and light green shading in the NIL sequence highlights the sequence ΔCS AATAGA.
- Fig.S3.jpg
Fig. S3 ChIRP-Seq analysis of Linc-GmSTT1 interacting with endogenous RNA and DNA in vivo. A, ChIRP analysis workflow. B, Venn diagram showing the overlapping RNAs peaks that interacted with Linc-GmSTT1 (three biological replicates). C, Venn diagram showing the overlapping DNA peaks that interacted with Linc-GmSTT1 (three biological replicates). D, Bubble plot showing the functional, enriched GO BP terms (left) and KEGG pathways of genes containing Linc-GmSTT1 RNA peaks (right). E, Bubble plot showing the functional, enriched GO BP terms (left) and KEGG pathways of genes containing Linc-GmSTT1 DNA peaks (right).
- Fig.S4.jpg
Fig. S4 Soybean lines used in this study: α-null hypoallergenic soybean near-isogenic line cgy-2-NIL (NIL) and α-normal control line (DN47). A, Backcross breeding process to generate the α-null NIL8. B, Genotype of NIL. The substituted region carrying the cgy-2 allele comprised 3.33 Mb. C, Representative NIL and DN47 plants and developing seeds collected at 20 DAF. The NIL and DN47 samples were phenotypically similar. D, The α-null mutation of β-conglycinin in NIL was verified by SDS-PAGE and western blot analyses. The α-null trait was stably inherited.
{kind=link}
{kind=link}
{kind=link}
{kind=link}