TGF-β induces EMT of A549 cells
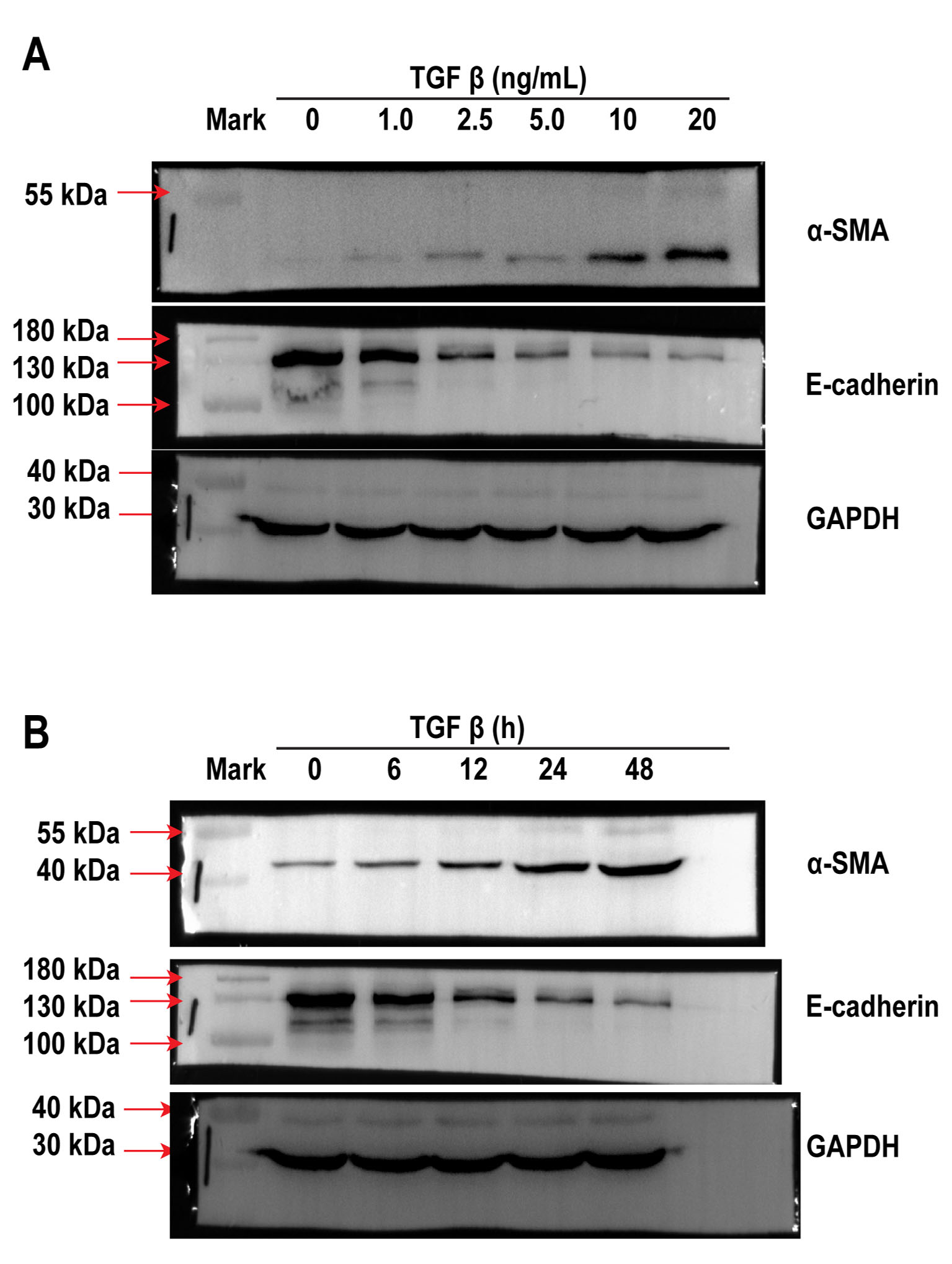
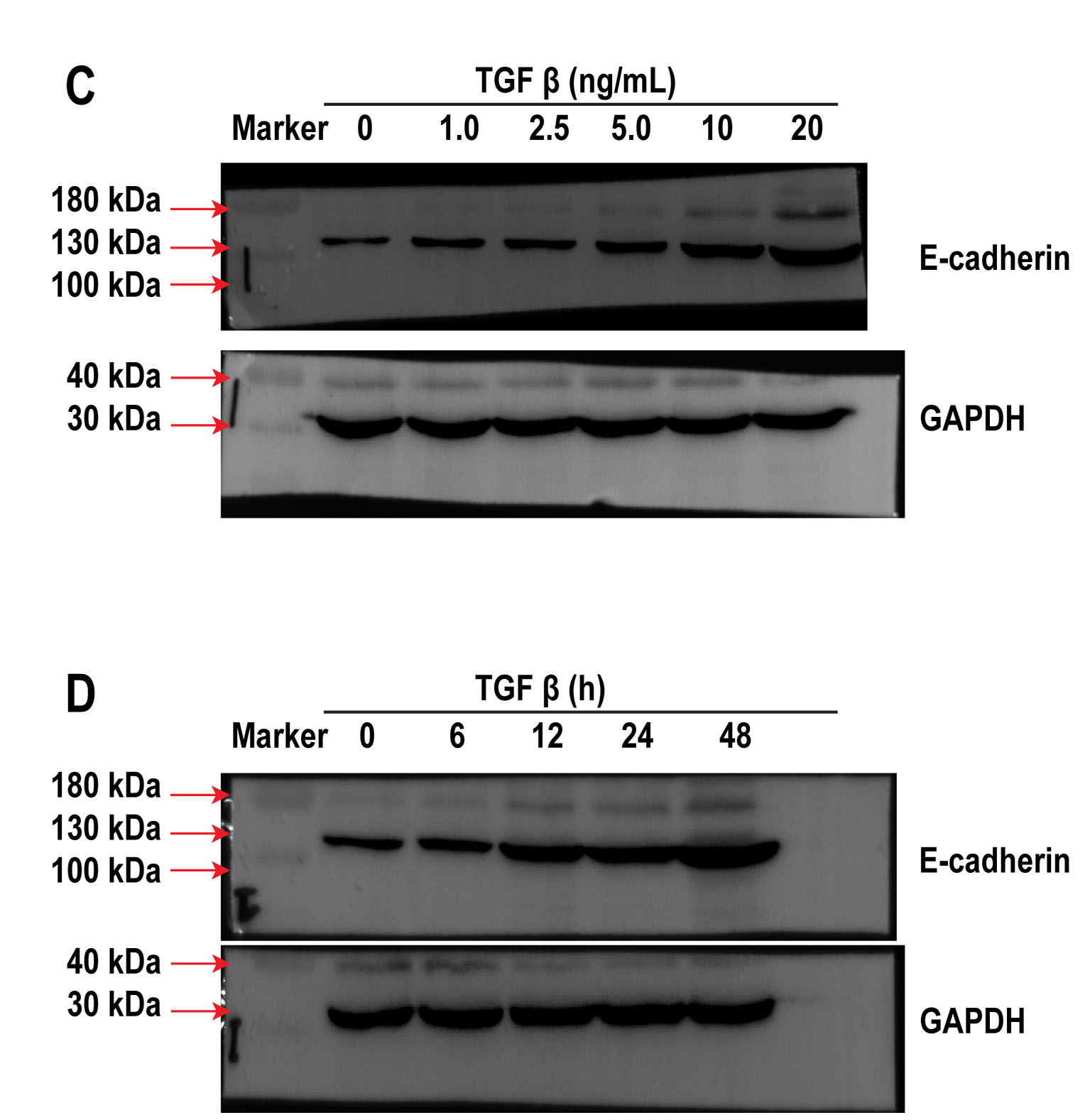
Based on previous work [23], the A549 cells were first treated with various amounts of TGF-β at different time periods to determine the safe and optimal concentration of TGF-β as well as the incubation time required for the investigation of EMT in vitro. When A549 cells were treated with 1.0, 2.5, 5.0, 10 and 20 ng/mL TGF-β for 24 h, we observed a dose-dependent increase in the expression of the mesenchymal marker α-SMA, while a decrease in the expression of epithelial biomarker E-cadherin was identified via western blotting (Fig.1A, S.1A), which collectively suggests mesenchymal differentiation. Although α-SMA level increased in A549 cells after treatment with 20 ng/mL TGF-β, cell viability was poor. In addition, A549 cells were treated with 10 ng/mL TGF-β at different time periods (0, 6, 12, 24, and 48 h) to determine the appropriate time frame of treatment. We observed an increase in the expression of α-SMA in a time-dependent manner, which peaked at 48 h by 8.72 ± 0.45-fold compared with that of the control, but cell death was evident. E-cadherin expression decreased significantly in a time-dependent manner after 24 h of exposure to TGF-β (Fig. 1B, S. 1B). Therefore, the optimal TGF-β concentration and incubation time were 10 ng/mL and 24 h, respectively. The level of collagen-I (determined using ELISA) increased upon A549 cells exposure to TGF-β (Fig. 1D), suggesting that an in vitro model of EMT-induced fibrosis was successfully established by treating A549 cells with optimal concentration of TGF-β for specific exposure duration.
Up-regulation of C/EBPβ is involved in TGF-β-induced EMT
Previous study has shown that phosphorylation of C/EBPβ is involved in pulmonary fibrosis in mice [24]. To investigate the precise roles of C/EBPβ in fibrosis, the relationship between C/EBPβ activation and TGF-β-induced EMT was assessed. In this study, we observed that TGF-β up-regulated C/EBPβ mRNA expression level in dose-and time-dependent manner (Fig. 2a, b). Furthermore, western blot analysis showed that the expression levels of C/EBPβ in A549 cells increased with dose-and time of TGF-β treatment at 10 ng/mL within 48 h (Fig. 2C, D, and S.2 C, D). The TGF-β-mediated increase in C/EBPβ expression was obvious when EMT was induced in the A549 cells. Next, we employed luciferase reporter assay to better understand C/EBPβ activation after TGF-β-induced EMT. Exposure of A549 cells to TGF-β generated time-and dose-dependent increase in C/EBPβ-luciferase activity and exhibited a 2.83 ± 0.42-fold increase in expression compared with that of the control (Fig. 2e, f). Collectively, these observations suggested that TGF-β increased the expression and activation of C/EBPβ during the EMT.
Loss of C/EBPβ shifts TGF-β-induced collagen deposition following EMT
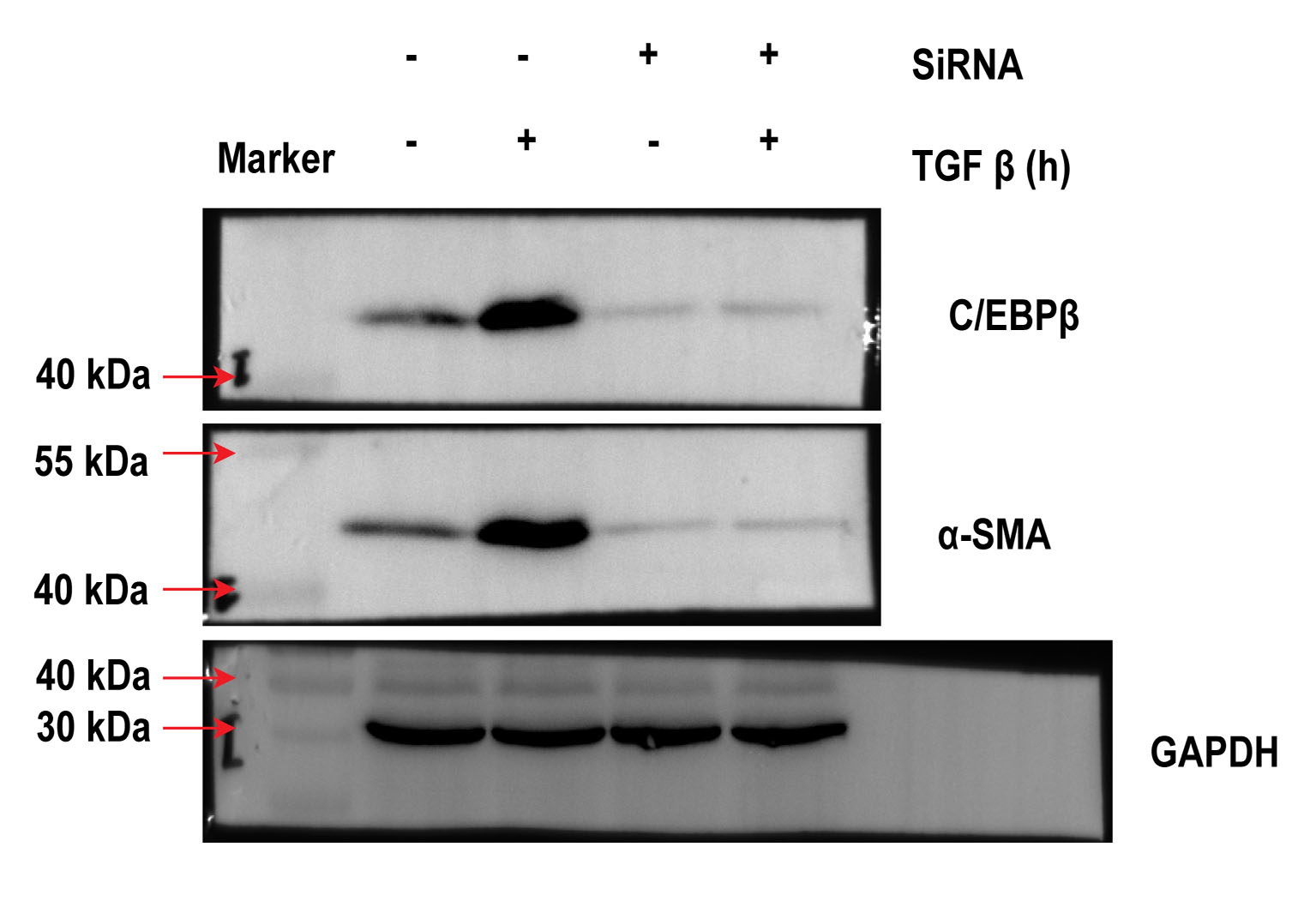
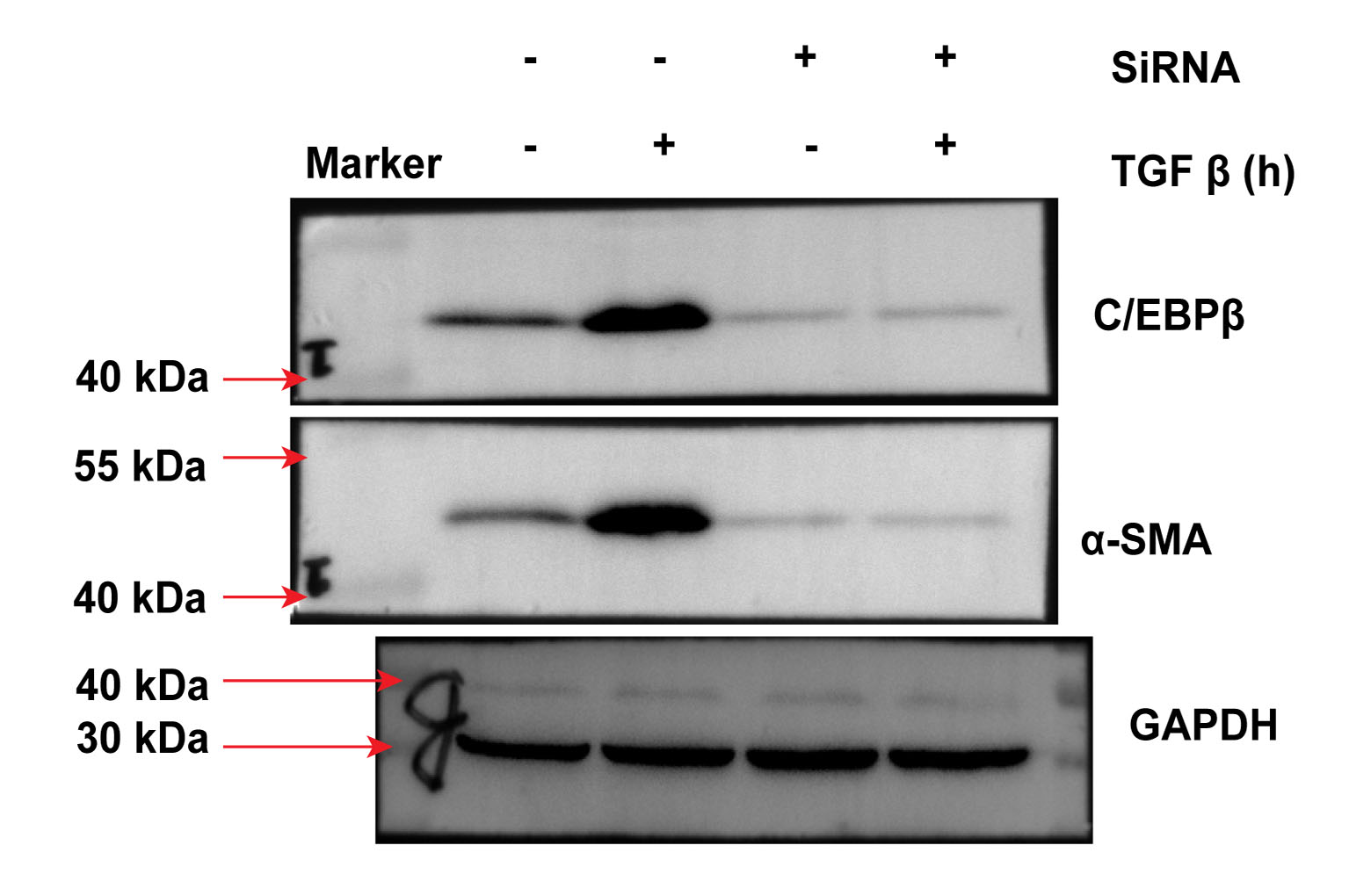
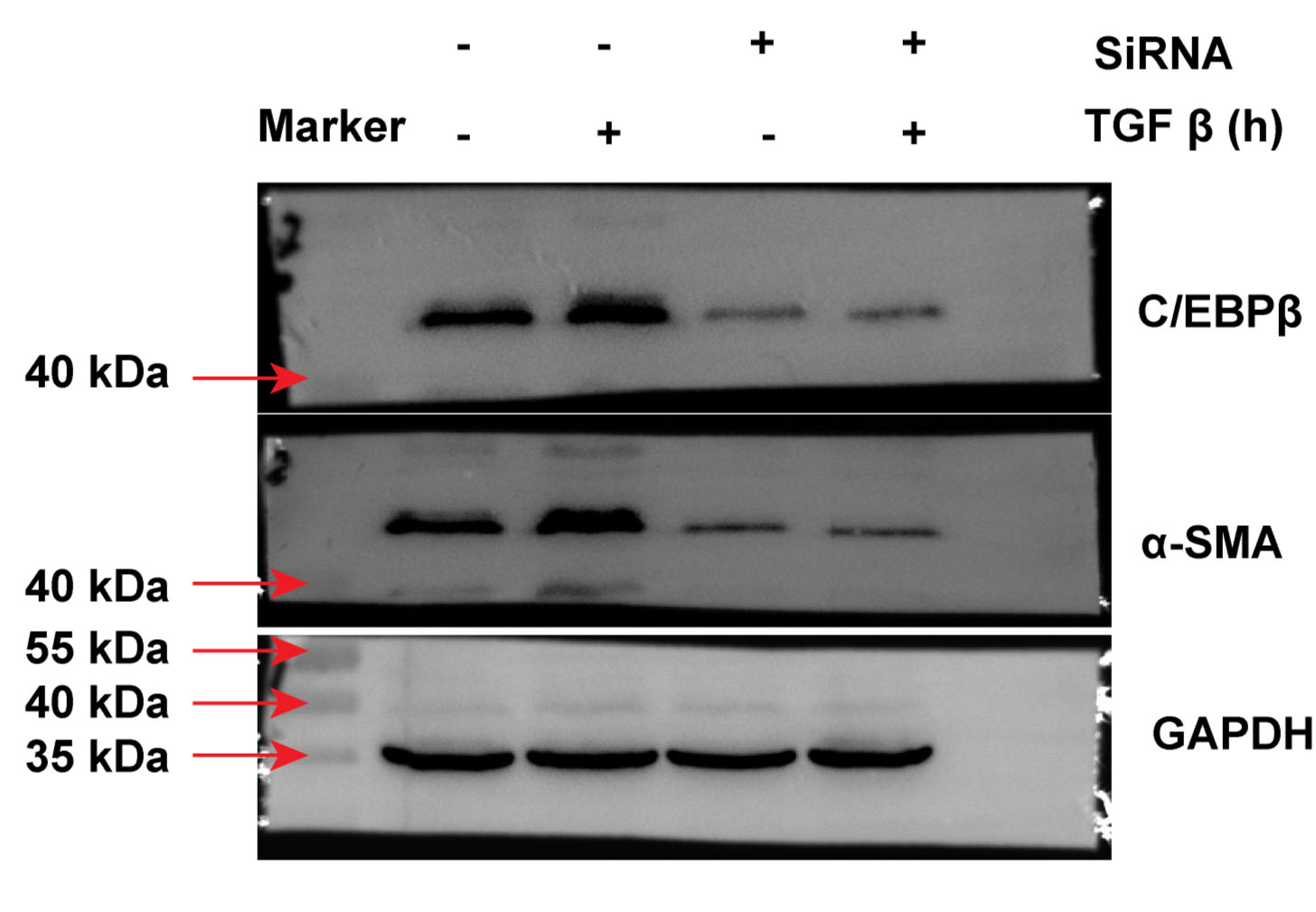
Previous studies have established the involvement of C/EBPβ activation in the pulmonary fibrotic process [15,16]. Other studies have also reported that mice with C/EBPβ deficiency antagonise BLM-induced pulmonary fibrosis in vivo [15]. In this regard, we investigated whether C/EBPβ is required for TGF-β-activated EMT and collagen-I deposition. Next, C/EBPβ siRNA (10 nM) was used to establish the reducing gene model in A549 cells. As shown in Fig. 3a, the C/EBPβ siRNA successfully decreased gene expression. Also, the A549 cells transfected with the C/EBPβ siRNA attenuated TGF-β-induced α-SMA and collagen-I expression (Fig. 3a, b, and S.4). Furthermore, TGF-β could not increase C/EBPβ-luciferase activity in A549 cells treated with the C/EBPβ siRNA (Fig. 3c). Taken together, our results suggest that C/EBPβ may be a crucial factor for the regulation of TGF-β-induced EMT and collagen-I deposition in pulmonary fibrosis.
TGF-β induced C/EBPβ binding to α-SMA promoter in A549 cells
As mentioned above, our data confirmed that C/EBPβ played a pivotal role in EMT and pulmonary fibrosis in vitro. As an important transcription factor, C/EBPβ triggered the expression of downstream genes by binding to its cognate sites in the gene promoters. However, the C/EBPβ binding site on the α-SMA promoter region in A549 cells is not known. Therefore, to elucidate the molecular mechanism through which TGF-β regulates α-SMA expression, we explored the putative C/EBPβ-binding sites in the 5′ promoter region of human α-SMA gene. Also, the 5′ promoter region of the human, mouse and rat α-SMA gene was examined via Multiple Sequence Alignment. We found a conserved putative C/EBPβ-binding motif TTGGGCAA in the 5′ promoter region within 200 bp from the identified transcription start site (Figure 4a. We therefore hypothesised that the putative C/EBPβ-binding motif was a C/EBPβ-responsive cis-element that mediates the upregulation of the α-SMA gene, and that the activation of this cis-element is critical for the development of EMT. To test our hypothesis, we evaluated C/EBPβ binding to the putative binding motif present in the α-SMA promoter in A549 cells through the ChIP assay. We observed that TGF-β-treated A549 cells showed increased C/EBPβ binding to the α-SMA promoter region (Fig. 4B, S. 4). Altogether, these results suggested that activated C/EBPβ could accelerate TGF-β-induced EMT by binding to the α-SMA promoter region in the A549 cells.
Role of acetylation of C/EBPβ in binding to the α-SMA promoter in A549 cells
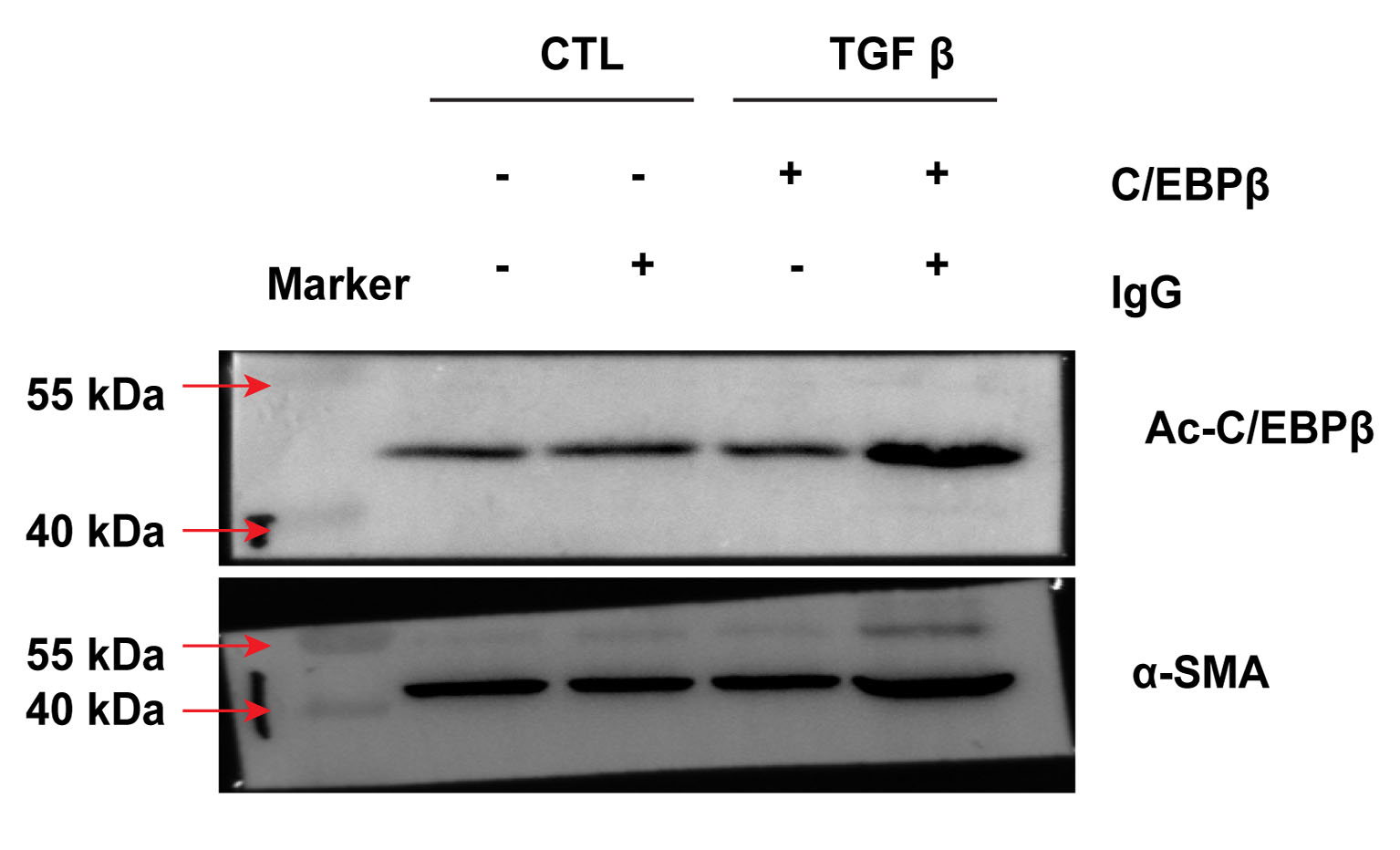
Other reports have shown that phosphorylation of C/EBPβ plays a critical role in alveolar EMT and acts an essential step in pulmonary fibrosis [25,26]. As a transcription factor, C/EBPβ activates its downstream signals via post-translational modification, such as phosphorylation, acetylation and methylation. However, reports on the role of C/EBPβ acetylation in pulmonary fibrosis are lacking. As shown above, activated C/EBPβ binds to the α-SMA promoter region in TGF-β-treated A549 cells. Nonetheless, the mechanism underlying C/EBPβ binding to α-SMA expression is not clearly understood. Hence, we investigated the effect of TGF-β on C/EBPβ modification and observed that acetylation of C/EBPβ could increase significantly by 6.42 ± 0.72 -fold in the A549 cells treated with TGF-β (Fig. 5, S. 5). An enhanced α-SMA expression was observed in samples treated with the C/EBPβ antibody but not in TGF-β-treated cells with immunoglobulin G (IgG). Collectively, our results showed that α-SMA expression may be triggered by C/EBPβ activation through acetylation.
Acetylation and deacetylation of C/EBPβ in TGF-β-induced EMT and collagen deposition
Herein, we used TGF-β-treated A549 cells to mimic the pulmonary fibrotic model in vitro. The binding of C/EBPβ to α-SMA coupled with the increased expression and acetylation of C/EBPβ indicated that acetylated C/EBPβ may be involved in EMT and pulmonary fibrosis. As demonstrated in Fig. 6a, TGF-β treatment led to acetylation of C/EBPβ and accelerated EMT-induced collagen-I deposition. Acetylation of C/EBPβ could be important for its activation. To clarify whether acetylation of C/EBPβ is necessary for pulmonary fibrosis, we subsequently investigated the effect of deacetylation of C/EBPβ on EMT using SIRT1. As reported, SIRT1, a class III histone deacetylase (HDAC), specifically deacetylates histone or non-histone proteins, while the C/EBPβ is one of the deacetylation targets of SIRT1 [27]. We observed that SIRT1 could reverse TGF-β-induced C/EBPβ acetylation (Fig. 6a). Importantly, C/EBPβ deacetylation significantly reversed the elevated expression of α-SMA and collagen-I in TGF-β-treated A549 cells (Fig. 6a, b). As indicated earlier in this work, SIRT1 stimulation also suppressed the increased C/EBPβ-luciferase activity in TGF-β-treated A549 cells (Fig. 6c). These observations suggested that acetylation and deacetylation are useful steps in regulating C/EBPβ functions. All these results confirm that C/EBPβ acetylation may be a key player in alveolar EMT and that pulmonary fibrosis is blocked by its deacetylation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}