2.1. Isolation and Characterization of ADSCs
This study was approved by the Animal Ethical Committee of Tarbiat Modares University, Tehran, Iran (IR.TMU.REC.1396.692).
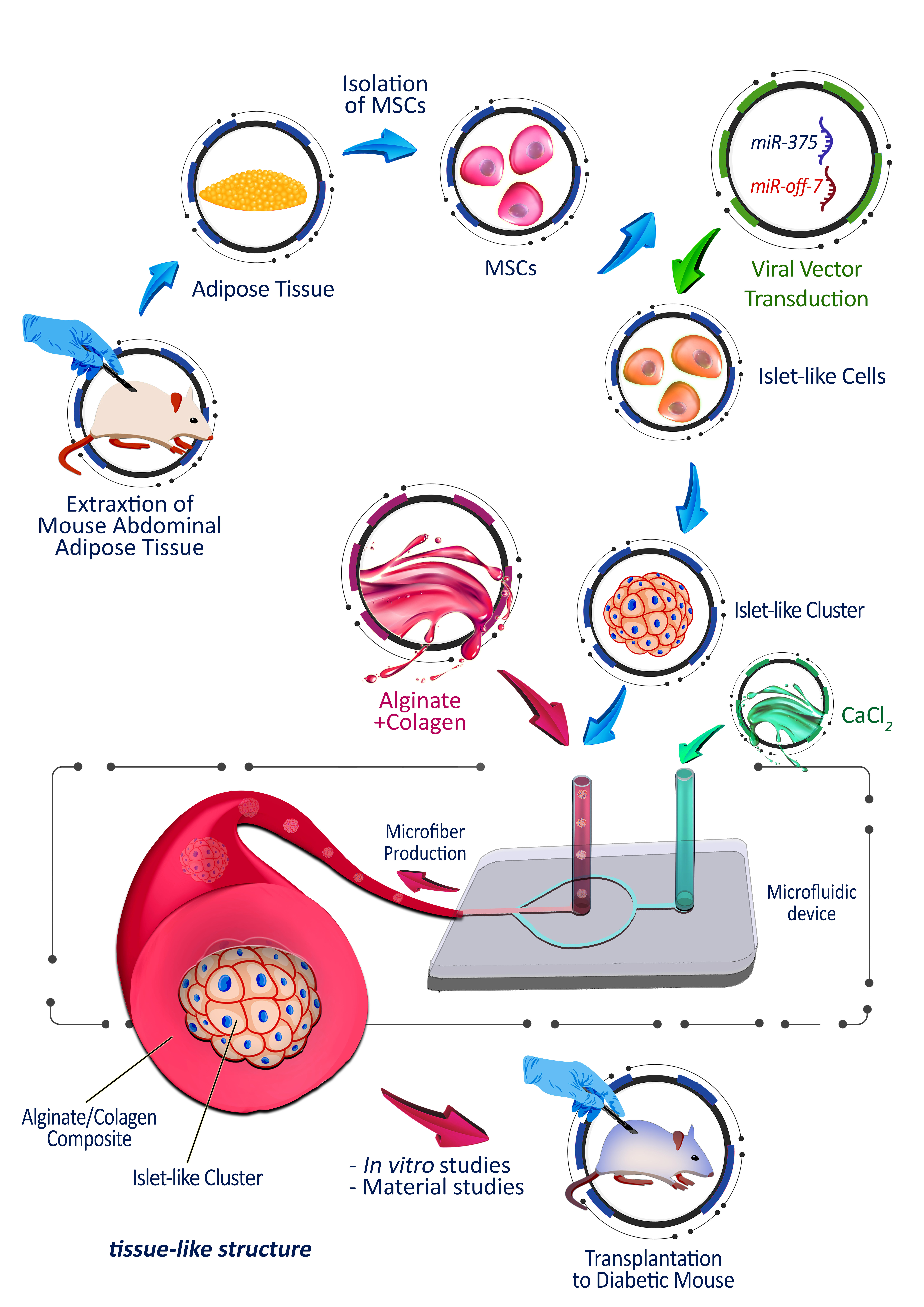
Adipose tissue was obtained from the abdominal cavity of Balb/c mice and digested for 45min at 37°C in PBS, pH=7.2 (Gibco, Germany), containing 2% BSA (Sigma-Aldrich, USA), and 0.2% collagenase type-І (Gibco, Germany). The isolated ADSCs at a density of 2×105 cells/cm2 were seeded into T25 culture flasks and incubated at 37°C, 5%CO2.
The ADSCs at passage three with appropriate monoclonal antibodies labelled with FITC or PE (BD Biosciences, USA) were analysed using fluorescence-activated cell sorting on a FACS Caliber (Becton-Dickinson, FAC scan, San Jose, CA, USA).
To confirm the adipogenic and osteoblastic differentiation potential of ADSCs, the cells were subjected to differentiation process and then, the cell were stained with Oil-Red-O and alizarin red (Sigma-Aldrich, USA) respectively and examined under phase-contrast microscopy (Olympus, Japan). The cells were also examined during osteogenesis for alkaline phosphatase activity using BCIP/NBT reagent (Becton Dickinson, Bioscience, UK) for 10–15 min [23].
2.2. Transduction of ADSCs with miR-375 and anti-miR-7 genes
The psPAX2 (containing gag and pol genes) and pMD2.G (containing VSV-G gene), as lentiviral packaging plasmids, mmu-mir-375 (containing the CMV and SV40 promoters (#abm. mm10408, Abm good. Canada), mmu-miR-7-inhibitor vector (#abm. MIM03178, Abm good. Canada) and pLenti-III-mir-GFP-Blank (#abm. m001, Abm good. Canada) were used for the co-transfection of HEK-293T cells. Plasmids then purified by midi Plus kit (MN, Germany) according to the instruction provided by the manufacturer.
HEK-293T cells (purchased from Stem Cell Technology Research Center, Tehran, Iran) were seeded in 6 cm Petri dishes with DMEM+10% FBS. The lentivirus carrying miR-375 and anti-miR-7 were propagated from the co-transfection in the HEK-293T cell line using the lipofectamin 2000 transfection reagent (Invitrogen, USA). The titer of the concentrated viral particles was determined using flowcytometry. To determine the titer of the GFP-expressing virus, a serial dilution was prepared and added to HEK-293T cells in culture. The transducing unit (TU/ml) of the GFP-expressing cells was determined for each dilution after 48 hours. A well containing HEK-293T cell without the viral dilution was also taken and considered as the negative control.
The ADSCs at passage three with 70–80% confluency were considered for lentivirus transduction. The experimental groups were designated as described below:
Group-1: The ADSCs transduced with mmu-miR-375 lentiviruses carrying GFP (ADSCs miR-375), Group-2: The cells transduced with mmu-miR-7-off lentiviruses carrying GFP (ADSCs anti-miR-7), Group-3: The cells infected with both miRNAs (ADSCs miR-375+anti-miR-7), Group 4: The cells infected with pLenti-empty lentiviruses carrying GFP (ADSCs null), Group 5: The control group, only ADSCs (no transduction).
The multiplicity of infection (MOI) of 30 was considered appropriate for all the experiments. The cells were cultured for one week in fresh complete serum-free medium containing DMEM supplemented with BSA (15%) and 2μg/ml puromycin (Sigma-Aldrich, USA).
2.3. Expression of miR-375, miR-7, and its gene targets in the transduced cells
The transduction efficiency was examined by GFP expression in the transduced cells under a fluorescence microscope. For determined the miR-375, miR-7 expression, total RNA was extracted from the cells using Trizol reagent (Invitrogen, USA) as instructed by the manufacturer, on day 4 of transduction. In following the experiment, total cellular RNA was isolated from transduced and control cells on days 7, 14, and 21 post-infection using Trizol for determined the pancreatic specific genes. The quality and quantity of the extracted RNA samples was checked on a Nanodrop (Thermofisher, USA). cDNA was synthetized from each RNA sample (2ng) using a cDNA synthesis kit according to the instructions given by the company (iNtRON Biotechnology, Korea). The expression of genes was determined using quantitative real-time PCR (QRT-PCR) using SYBR green kit (Takara, Korea). The assay was performed in triplicate in a reaction mixture on a real time PCR system (Applied Biosystems, ABI-7500). PCR reaction was performed with mmu-miR-375, mmu-miR-7 primers, and U6 small nuclear RNA endogenous control primers. The qRT-PCR cycling condition was as follows: 95 C for 10 min, followed by 40 cycles (each cycle for 15 sec at 95 C and 1 min at 60 C). The primers used in this experiment are as listed in Additional file 1: Table S1. The relative quantification (ΔΔCt) method was applied to calculate the data. Using this assay the gene transcripts of IPCs were compared with that of a mouse pancreatic beta-cell line (MIN-6, purchased from Iranian Biological Resource Center, Tehran, Iran).
2.4. Visualization of the spheroid IPCs and immunofluorescence staining
Formation of the spheroid IPCs obtained on day 21 of differentiation from ADSCs was evaluated by dithizone (DTZ) staining [24]. The cells were further characterized by showing specific protein localization by ICC technique [25]. The IPCs were treated with primary antibodies against Insulin (#ab7760, Abcam, Cambridge, MA, UK), Glucagon (#ab10988, Abcam), Pdx1 (#ab84987, Abcam), and Neurogenin3 (#ab87108, Abcam). The FITC-coupled goat anti-mouse IgG (#AF8032, Razi Biotech, Iran) and Texas Red-labelled goat anti-mouse IgG (#ab175473) were used as secondary antibody. Besides, samples were processed without primary antibody and considered as negative control. All the ICC assays were performed in triplicate. Wherever indicated, the nuclei of the cells were stained with DAPI (0.1μg/ml).
2.5. Estimation of insulin and C-peptide secretion by IPCs
Insulin and C-peptide levels secreted by the IPCs were measured using an ultrasensitive mouse ELISA kit (#Mecodia, Uppsala, Sweden, and # Alpco, Salem, USA respectively). The assays were carried out according to the procedure described by the manufacturer’s instruction. Glucose-stimulated insulin release was assayed in differentiated cells after the cells were incubated for 2 hours in freshly prepared Krebs-Ringer bicarbonate buffer (KRB) (Sigma-Aldrich, USA) without glucose. Then the cells were incubated for 2 hours in KRB containing 0.5mmol IBMX (Iisobutyl- 3-methylxanthine (Sigma-Aldrich, USA) and different glucose concentrations (5, 10, 15, 20, 25, and 30 mmol). In case of insulin and C-peptide assays in the cells, the cells were first washed for three times with PBS, extracted in 0.2ml acid alcohol (10% glacial acetic acid in absolute ethanol) at 4°C overnight, and sonicated briefly before centrifugation at 3500 g for 15 min at 4°C. Total protein concentration was determined by BCA protein assay system and 50μg of protein was used for detection of intracellular insulin and C-peptide in each well of ELISA kit. Absorption was recorded at 450nm and all the assays were carried out in triplicate.
2.6. Assembly of microfluidic device and Collagen-Alginate micro-fiber
In this experiment, microfluidic device was fabricated using standard soft lithography methods [26, 27]. Cylindrical and coaxial-flow channels were prepared by aligning and bonding Poly dimethyl siloxane (PDMS) channels by oxygen plasma treatment (Harrick Scientific, Ossining, NY) to produce Collagen-Alginate fibers. The diameters of the cylindrical channel at the inlet, tapered junction, and outlet were 400, 200, and 600 μm, respectively. In this device, solutions were supplied from the inlets: a sample fluid (cells suspended in Collagen-Alginate solution), and a sheath fluid (3% (w/v) calcium chloride, dissolved in deionized water). Sodium alginate (Sigma-Aldrich) was prepared in culture medium and collagen by mixing collagen solution and alginate and gently pipetting with cell pellets at a concentration of 1.5×106 cells/ml. The flow rate of each fluid was adjusted to 100μl/min with the help of a syringe pump and the sheath flow rate was about 1ml/min. The extruded fibers were collected in a Petri dish containing CaCl2 and incubated in culture medium at 37˚C for 45min.
2.7. Characterization of the microfiber
Live/dead staining: The viability of encapsulated IPCs was evaluated at 1 and 7 days after encapsulation using mixture of acridine orange (100 μg mL−1) and ethidium bromide (100 μg mL−1) and viewed using a fluorescence microscope. Acridine orange penetrates into cells and stains the DNA of live cells green fluorescence using blue filter, whereas ethidium bromide only enters dead cell membranes, stains the DNA of dead cells using green filter and generates a red fluorescence. Two images taken from the same field were merged by Photoshop8 CS software.
Cytotoxicity assessment: Cytotoxicity of the Collagen-Alginate was assessed using 3-(4, 5- dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. The differentiated encapsulated cells were soaked in DMEM supplemented with 5% FBS. The cell proliferation was evaluated during 14 days in Collagen-Alginate fiber. Briefly, culture medium was removed and replaced with 20μl MTT (0.5mg/ml) (Sigma) was added to each well and plates were incubated at 37°c in dark. The assay was carried out in triplicate, and the results are presented as mean ±SD.
Microstructural of fiber (SEM): The surface of the freeze-dried fibers was examined using SEM (Philips XL30, Amsterdam, Netherlands). The porosity of fibers was measured using ImageJ software.
Fourier Transform Infrared Spectroscopy (FT-IR): The chemical structure of the synthesized hydrogels was investigated by using FT-IR spectroscopy (FT-IR 8400S, Shimadzu, Japan) in the wavenumber of 400–4000 cm−1.
Swelling property was examined by doing the cross-linked composite porous fibers.
The fibers were weighed every 50 min and then immersed in excess of swelling medium (pH=7.4) at 37◦C until they reach the equilibriums. At various time intervals, the hydrogel was removed from the solution and weighed after blotting the excess water using blotting paper. The weight of the swollen fibers was recorded every 50 min until they reach equilibrium state. Data presented in this paper are mean values of triplicate measurements. The percentage swelling of fibers was calculated according to the following equation: Q = (Ms – Md) / Md. Where, Q is the swelling ratio, Ms is the mass in the swollen state and Md is the mass in the dried state.
2.8. In vivo experiment
This experiment was carried out on male BALB/c mice. The mice were 8-10 weeks-old weighing 30±5g. Animals were housed in standard cages, with food and water ad libitum, under controlled temperature condition (23 ± 2 °C). The experimental groups are as follows; Group-1: Diabetic mouse received IPCs (n=5). Group-2: Diabetic mouse received MSCs (n=5). Group-3: Non-diabetic mice l (n=5). Group-4: diabetic mice as Control (n=5).
Wherever indicated Type1 diabetes was induced in mice by administration of streptozotocin (STZ) treatments. Each mice received intra peritoneal (I.P) four consecutive injections of STZ (80mg/Kg dissolved in citrate buffer, pH=4.5). Blood glucose level (non-fast) was readily monitored using a portable glucometer on blood samples collected from the tail vein (Accu-CHEK, Roche). Blood glucose was measured at different time intervals and considered as an index for hyperglycemia (diabetes). Blood glucose above 17mmol/l was considered as abnormal which was achieved 5 days after the STZ administration. The animals were selected for transplantation received approximately 1.5×106 differentiated and non-differentiated cells encapsulated by the Collagen-Alginate fibers. Each mouse received subcutaneous implantation of fiber-entrapped cells on the back in the anaesthetized status using 50 mg/kg ketamine and 5 mg/kg xylazine.
The functional efficacy of the transplant was evaluated after four weeks as shown by glucose tolerance test (GTT). Change in blood glucose in fasting mice was monitored for 6–10 hours at different time intervals (20, 40, 60, 90 and 120 min) following treatment with 2g glucose/kg body weight injected I.P. After 5 weeks of post transplantation (at the end of the treatment period before sacrificing), animals were anesthetized with an I.P. injection of ketamine (70 mg/kg) and xylazine (10 mg/kg) and 2ml of blood sample was collected by heart puncher and used to measure insulin level.
2.9. Statistical analysis
The statistical analysis was performed using One-way ANOVA and Bonferroni’s post hoc test by Graph pad Prism5 software (GraphPad Software Inc., La Jolla, CA). The results are presented as mean ±SD and P-value less than 0.05 was considered statistically significant.
{kind=link}