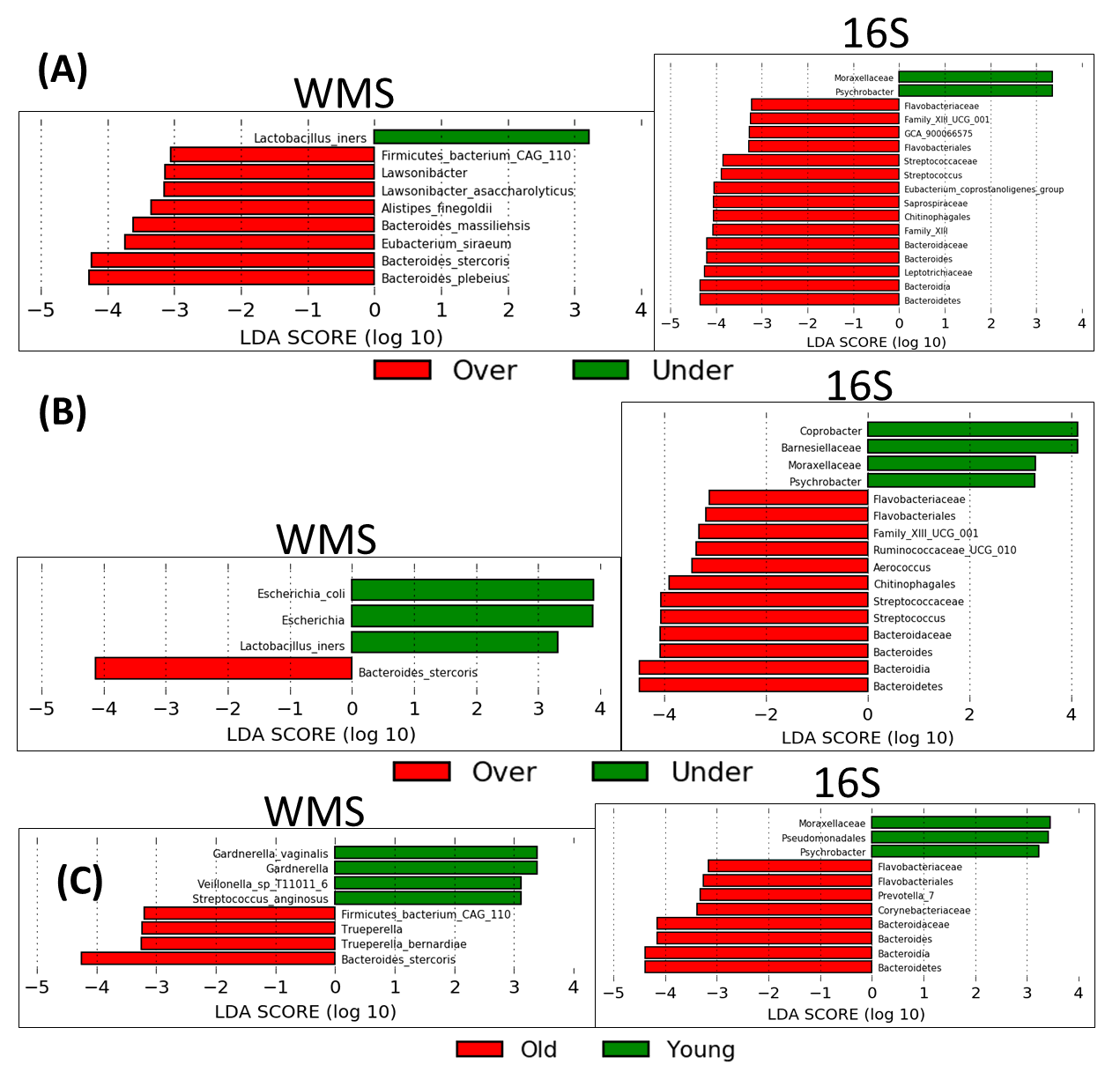
This study is limited by our analytic pipelines and available samples, but the results suggest that 16S with OTU clustering provides a similar description of sample diversity and composition for gut microbiomes of cervical cancer patients versus WMS. This finding is important, as it allows researchers to analyze a larger number of samples using 16S at a fraction of the cost of WMS. Camargo evenness and skewness were the least correlated indices between the two methodologies, which suggests that the sequencing methods differ in terms of the proportionality of individual bacterial taxa. This might be improved using a 16S analysis pipeline that uses amplicon sequence variants, such as QIIME2, to retain more reads. The Camargo index has low sensitivity for variation in species diversity for sample sizes < 3000, while the Pielou index is a sensitive assessment index for smaller sample sizes (< 1000). Thus, the Pielou evenness index is more appropriate in terms of this sample size, and correlates well between the two datasets[20]. With regards to rare taxa (LAR), WMS provides more noise in a dataset by identifying individual genes, which may be linked to unidentified bacterial species. 16S combined with OTU clustering can at best provide information at the genus level with a high degree of confidence and relies on 97% similarity clustering at the OTU level. This difference is to be expected, and could be exploited in specific analyses, such as searching for previously identified species or particular gene functions. It is reassuring that there was significant consensus between the methodologies on the higher order levels. Much of the focus in next generation sequencing analysis is placed on the smallest taxonomic level available (i.e. the genus or species level), but higher order taxa also provide valuable information.
Previous work has also posited a sizable amount of agreement between 16S and WMS sequencing techniques at higher orders of taxa[2], consistent with these results. 16S and WMS have a significant degree of correlation; however, most of those studies utilize data derived from samples collected in similar but not identical contexts. This project provides a unique opportunity in that both 16S and WMS sequencing datasets were derived from a single sample collected from each patient and then bacterial DNA was extracted for both methods. Using this high-quality information, we investigated the correlation of these two datasets in terms of microbial composition abundance and alpha diversity, to precisely determine how well these sequencing methods corroborated. Since the two datasets are derived from the same samples, association with the clinical variables should also result in the same conclusion regardless of the sequencing method used, which was again confirmed. Age is perhaps the variable most strongly associated with microbiome diversity, which was confirmed in both datasets in our study. It is also important to note, hierarchical clustering analysis showed 9 (69%) out of 13 patients in Cluster1 were white, while only 8 (31%) of the 28 patients in the rest of the cohort were white. In addition, 12 (92%) out of the 13 patients in Cluster1 had a disease stage of 1 or 2, compared to 19 (68%) out of the 28 patients in the rest of the cohort.
An important limitation of the study that we focus solely on taxonomic characteristics of the gut community. The major advantage of WMS is that it provides an opportunity to assay functional diversity of the microbiome, a capability severely lacking in 16S data. Tools such as PICRUSt[26] can infer metabolic profiles from 16S data, but they cannot truly assemble functional pathways. Yet, the most fundamental drawback of this study is due to the limitations of analytic pipelines used in each approach and the databases available for both 16S and WMS data. Tools for analyzing 16 s have been developed and successfully deployed far longer than WMS analysis software, while the WMS analysis pipelines and databases are continually being developed and shared. The differences in alignment techniques and databases would account for a lot of the variation in taxa names herein. For example, by calculating OTUs we recapitulated a popular method of alignment used in this field, but in doing so the data has been collapsed at the cost of potential diversity information. Additionally, tools used for metagenomic analysis vary based on techniques used such as distance metrics and clustering approaches[15]. Here, we used OTU clustering at 97% similarity using previously described methodology from the Human Microbiome Project[13], but this data could be re-analyzed using QIIME2 and amplicon sequence variant (ASV) calling[27] and result in variations in ASV vs. OTU assignment that could affect the analysis. Amplicon sequence variant calling with DADA2 denoising[28] may be a preferable system for WMS comparisons as the pipeline is more similar to how WMS reads are treated. Another important consideration is that the MetaPhlAn2 tool inherent in the Humann2 pipeline uses a relatively small fraction of the data generated, whereas another non-marker gene based identifier such as QIIME2, Kraken 2[29] or the mothur software[30] will generate a larger and more varied, spread of results. Still, MetaPhlAn2 outperformed IGGsearch[31] which was also deployed on our WMS dataset, and it remains the most popular marker-gene based tool in the metagenome field.
Another limitation to address, for this work and many others, is establishing a confident rarefication cut off for analysis. Usually this cut off value would be validated by utilizing a mock microbial community dataset to be analyzed alongside experimental data. Here, we were unable to acquire complete mock communities as such information is privileged and difficult to attain. However, the cut off value we used was selected because it was consistently stringent across both 16S and WMS datasets while retaining as much information as possible.
All this is to say, variations in approaches to metagenome assembly pipelines similarly could affect taxonomic assignment in 16S and WMS data. It is possible that a particular sequence relevant to both datasets would be classified differently during preprocessing, highlighting the necessity of universal reference databases and sequencing alignment tools and protocol consensus.
Given this variability in sequencing and data processing pipelines, the use of multiple techniques across different types of sequencing data is an excellent way to confirm consistency in conclusions. However, limited resources (e.g. material from clinical samples, bioinformatics support, time and finances) hamper the ability for this expansive and in-depth microbiome profiling for all studies. Although WMS has been demonstrated to confer significant advantages over 16S, this work suggests there is very little additional taxonomic information identified from WMS that was not identified in 16S data. This can vary depending on the context of analysis, for example method of sample collection (i.e., whole stool vs swabs) and determining the functional components of the microbiome in question.[26, 32] It is even possible that extracting DNA from the same sample at two different times, instead of splitting a single extraction as was done here, may yield slightly different results. In our work, alpha diversity assessments such as overall diversity, evenness and richness can provide meaningful, and more important, comparable information (Fig. 1) when obtained with either 16S or WMS. Furthermore, the two datasets provided a high degree of consensus when these indices were subjected to statistical analysis. This suggests that for studies where overall microbiome diversity, richness and evenness are the goals of an analysis, 16S is more than sufficient to provide this information. For basic taxonomic descriptions, there was a meaningful agreement on the phyla and higher taxa levels, suggesting that 16S is also sufficient in this setting for hypothesis-generating data. Nonetheless, these two datasets did provide some differences in taxonomic assignment, particularly on the genus level, and relative abundances of individual taxonomies. This suggests that for studies where a broader repertoire of potential species are needed, both techniques may be necessary.
{kind=link}