RNA sequence data analysis
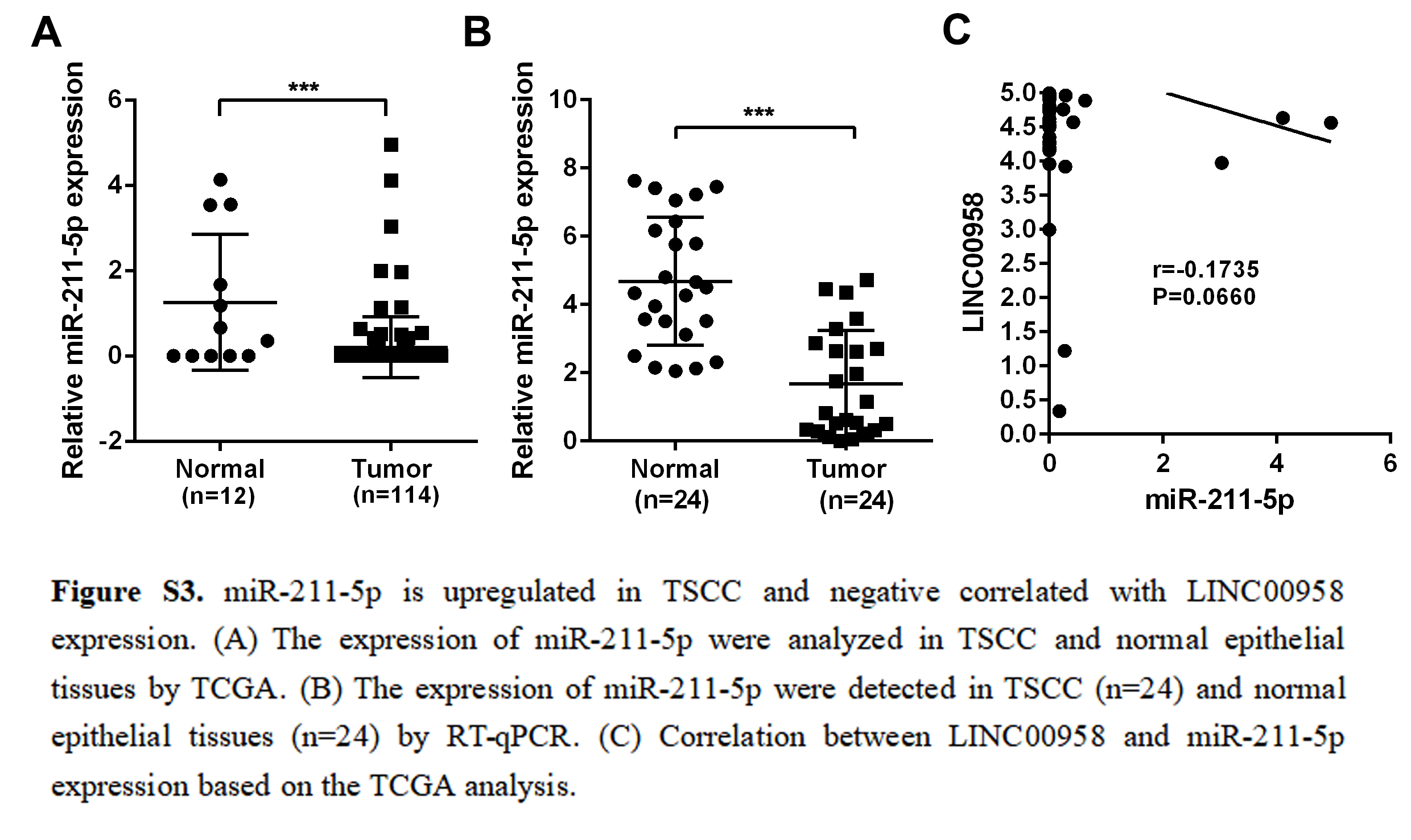
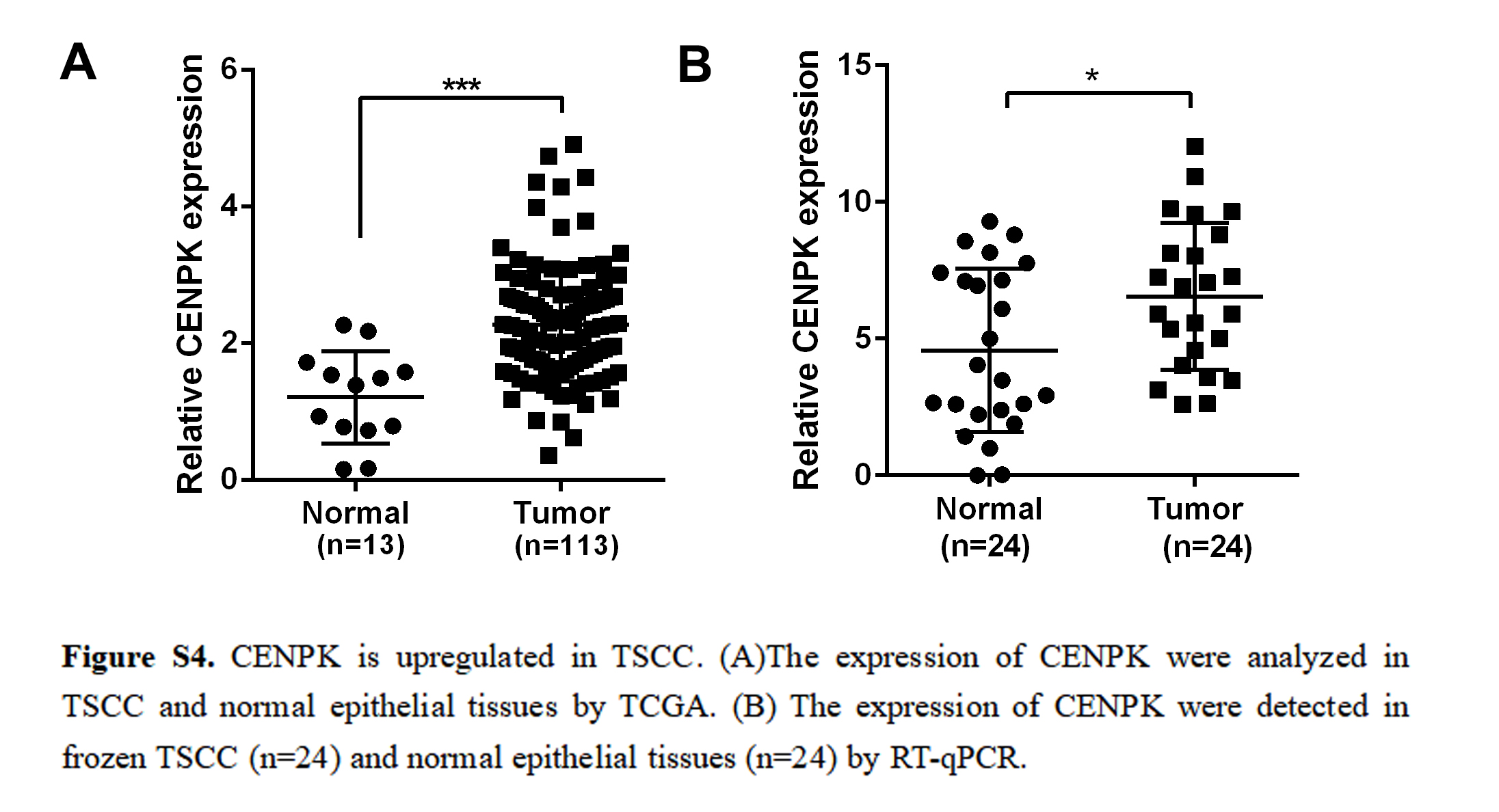
RNASeq and miRNASeq data from TSCC samples were downloaded from the Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/). All the data are publicly available. lncRNAs were identified according to the Ensembl database (http://www.ensembl.org/index.html, version 89) lncRNAs that were not included in this database were excluded. The edgeR package (Robinson, McCarthy & Smyth, 2010) was used to normalize gene expression of mRNAs (DEmRNAs), miRNAs (DEmiRNAs), and lncRNAs (DElncRNAs) in TSCC and normal tissues. Absolute log2FC ≥ 2 and FDR < 0.01 were used as cut-off criteria. Survival R package were further used to exclude the differentially expressed lncRNA without OS at P-value < 0.05. Next, the screened lncRNAs were used to predict lncRNA-miRNA interactions according to TargetScan (Fromm et al., 2015). lncRNAs included in these interactions were used for further study.
Clinical specimens
Patients with TSCC, who were diagnosed, treated, and followed up at the Department of Oral Surgery, Stomatological Hospital, Southern Medical University, were included in the study. This study was approved by the hospital institutional review board and written informed consent was obtained from all the patients. All the protocols were reviewed by the Joint Ethics Committee of the Southern Medical University and performed following national guidelines. Tissue samples were collected at surgery, immediately frozen in liquid nitrogen, and stored until total RNA or proteins were extracted.
Cell Lines and Culture
HOK and five TSCC cell lines (CAL-27, SCC-9, SCC-4, SCC-15, SCC-25) were obtained from the Tumor Cell Bank of the Chinese Academy of Medical Science (Shanghai, China). CAL-27 cells were maintained in DMEM medium (Gibco, Grand Island, USA), which was supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, USA), and the other cells were cultured in RPMI-1640 (Gibco, Grand Island, USA) supplemented with 10% FBS. For all cell lines, 100 IU/ml penicillin and 100 μg/mL streptomycin were added to the culture medium, and all of the cells were incubated at 37℃ in a humidified atmosphere of 95% air 5% CO 2.
Real-time quantitative PCR (RT-qPCR)
Total RNA was isolated from cells or tissues using TRIzol reagent (Invitrogen, California, USA) and then was converted to cDNA using a PrimeScript RT reagent kit (TaKaRa, Tokyo, Japan). RT-qPCR analysis were carried out in triplicate for each sample using SYBR Green Master Mix (TaKaRa, Tokyo, Japan). All primers are listed in Table S1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the endogenous control. For detecting miRNA expression level, cDNA was synthesized using MicroRNAs qPCR Kit (SYBR Green Method) (Sangon Biotech, Shanghai, China), and U6 small nuclear RNA served as the endogenous control.
CCK-8 assay
Cell viability was determined using the CCK-8 assay. Briefly, 2×103 cells/well were seeded into 96-well plates, and the absorptions of the cells were measured using a CCK-8 kit (Dojindo, Kyushu, Japan) according to the manufacturer’s instructions at different indicated time points. Data were from three separate experiments with four replications each time.
Clone formation assay
From each group, nearly 1×103 cells were plated in each well of a 6-well culture plate. Each cell group consisted of three wells. The cells were incubated at 37 °C for 14 days with growth media being replaced every third day. Then, the cells were washed twice with PBS and stained with 0.5% crystal violet.
EdU Incorporation Assays
Cells were cultured in 24-well plates, and 10 μM EdU was added to each well. Then, the cells were cultured for 2 h at 37°C and were fixed with 4% formaldehyde for 20 min at RT. After washing with PBS, the incorporated EdU was detected with a kFluor488-EdU kit (KeyGEN, Jiangsu, China) for 30 min at RT, and subsequently, the cells were stained with Hoechst 33342 for 20 min and were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and were merged using Adobe Photoshop 6.0 software. All experimental procedures were repeated at least three times.
Cell cycle analysis by flow cytometry
Cell cycle analysis were performed using the Cell Cycle Analysis Kit (Beyotime, Jiangsu, China) as per the manufacturer’s instructions. Cells were harvested and fixed in 70% ethanol overnight at 4 ℃. Then, the cells were stained with 25 μg/mL propidium iodide containing 1 μg/mL RNase at 37 ℃ for 30 min. The cells were analyzed for their distribution in different phases of the cell cycle on FACSCalibur flow cytometer using CellQuestPro software (Becton Dickinson, New Jersey USA).
RNA immunoprecipitation (RIP)
A Thermo Scientific RIP kit (Thermo, Waltham, MA, USA) was used to carry out RIP according to the manufacturer’s instructions. CAL-27 and SCC-9 cells were transfected with miR-211-5p mimics or mimics NC. Complete RIP lysis buffer was used to lyse cells. Magnetic beads conjugated with anti-Argonaute 2 (AGO2, Millipore, Massachusetts, USA) or control anti-immunoglobulin G (IgG, Abcam, Cambridge, England) antibody were used to incubate the cell extract. The cell extract was incubated for 6 h at 4℃. As the protein beads were removed, RT-qPCR was conducted for the purification of RNA.
Western blot analysis
Cell lysates was separated by SDS-polyacrylamide gel (4%–10%) electrophoresis, and then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Massachusetts, USA). The membranes were then blocked with 5% skimmed milk and incubated overnight at 4°C with the following primary detection antibodies: anti-CENPK (1:1000, Abcam, Cambridge, England), anti-C-MYC (1:1000, Abcam, England), anti-Cyclin D (1:1000, CST, Boston, USA), anti-Cyclin E (1:1000, Abcam, Cambridge, England), anti-Rb (1:1000, CST, Boston, USA), anti-p-Rb (1:1000, CST, Boston, USA), anti-GAPDH (1:10000, Abcam, Cambridge, England), anti-JAK1 (1:1000, Abcam, Cambridge, England), anti-STAT3 (1:1000, Abcam, Cambridge, England), or anti-p-STAT3 (1:1000, Abcam, Cambridge, England). The species-matched secondary antibodies were then incubated for 1 h at room temperature and the proteins were detected using BeyoECLPlus (Beyotime, Jiangsu, China).
Luciferase reporter assay
The wild-type or mutant LINC00958 fragment or CENPK 3'-UTR containing the predicted binding sites of miR-211-5p. were subcloned into a psiCHECK2 Dual-luciferase vector (Promega, Madison, USA). The luciferase reporter plasmids were co-transfected into TSCC cells with miR-211-5p mimics or the negative control. The relative luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega, Madison, USA) according to the manufacturer’s instructions.
IHC staining
We used anti-Ki67 antibody (CST, Boston, USA), anti-PCNA antibody (Abcam, Cambridge, England), anti-CENPK antibody (Abcam,Cambridge, England) and anti-p-STAT3 (CST, Boston, USA) to detect the expression in the mouse xenografts. The sections were visualized using a Nikon ECLIPSE Ti microscope system and were processed with Nikon software.
Plasmids, Virus Production, siRNA, and Transfection
The full length of LINC00958 and shRNAs targeting LINC0058 was synthesized and cloned into the lentiviral plasmid pSin-EF2-puromycin (Addgene, Cambridge, MA, USA). pSin-EF2-LHX2-puromycin or negative control pSin-EF2-puromycin vector was then co-transfected into 293T cells with the VSVG and PSPAX2 packaging plasmid (Addgene, Cambridge, MA, USA) using Lipofectamine 3000 reagent (Invitrogen). The supernatants were harvested and used to infect CAL-27 and SCC-9 cells, and stable clones were selected using 0.5 μg/mL puromycin. Small interfering RNA targeting CENPK (si-CENPK) was synthesized by RiboBio (Guangzhou, China). For CENPK knockdown, CAL-27 and SCC-9 were transfected with si-CENPK (50 nM) with Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s instruction and harvested for assays (48 h after transfection). miR-211-5p mimics, miR-211-5p inhibitors, and miR-controls were purchased from RiboBio (Guangzhou, China). MiRNA mimics, miRNA inhibitors, and miR-controls were transfected into cells at a concentration of 50 nM using Lipofectamine 3000 (Invitrogen, California, USA).
In vivo nude mouse models
A total of twenty-four 5-week-old male nude mice were purchased from Guangdong Medical Laboratory Animal Center, and kept under specific pathogen-free conditions. Mice were divided into four groups at random and inoculated with Cal-27 and SCC-9 cells subcutaneously on the right flank with 2 × 106 cells (n = 6 per experimental group). The growth of the tumors was observed and the volumes of tumors were measured every 4 days. The animals were sacrificed after 4 weeks of growth, and the tumors were excised for pathological examination.
Subcellular Fractionation and Fluorescent in Situ Hybridization (FISH) assay
Nuclear and cytoplasmic RNA was separated with the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Invitrogen) accordingly, and then analyzed by quantitative RT-PCR. FISH was performed for subcellular localization of LINC00958. The cover glasses were placed into a 24-well plate and cells were inoculated at the density of 6×104 cells/well. When cell confluence reached 80 %, the cover glasses were removed, and the cells were washed by PBS and fixed with 1 mL 4 % paraformaldehyde at room temperature. The experiment was conducted according to the instructions of RiboTMlncRNA FISH Probe Mix (Red) (RiboBio Co., Ltd., Guangdong, China). The cells were observed and photographed under a fluorescence microscope.
Statistical Analysis
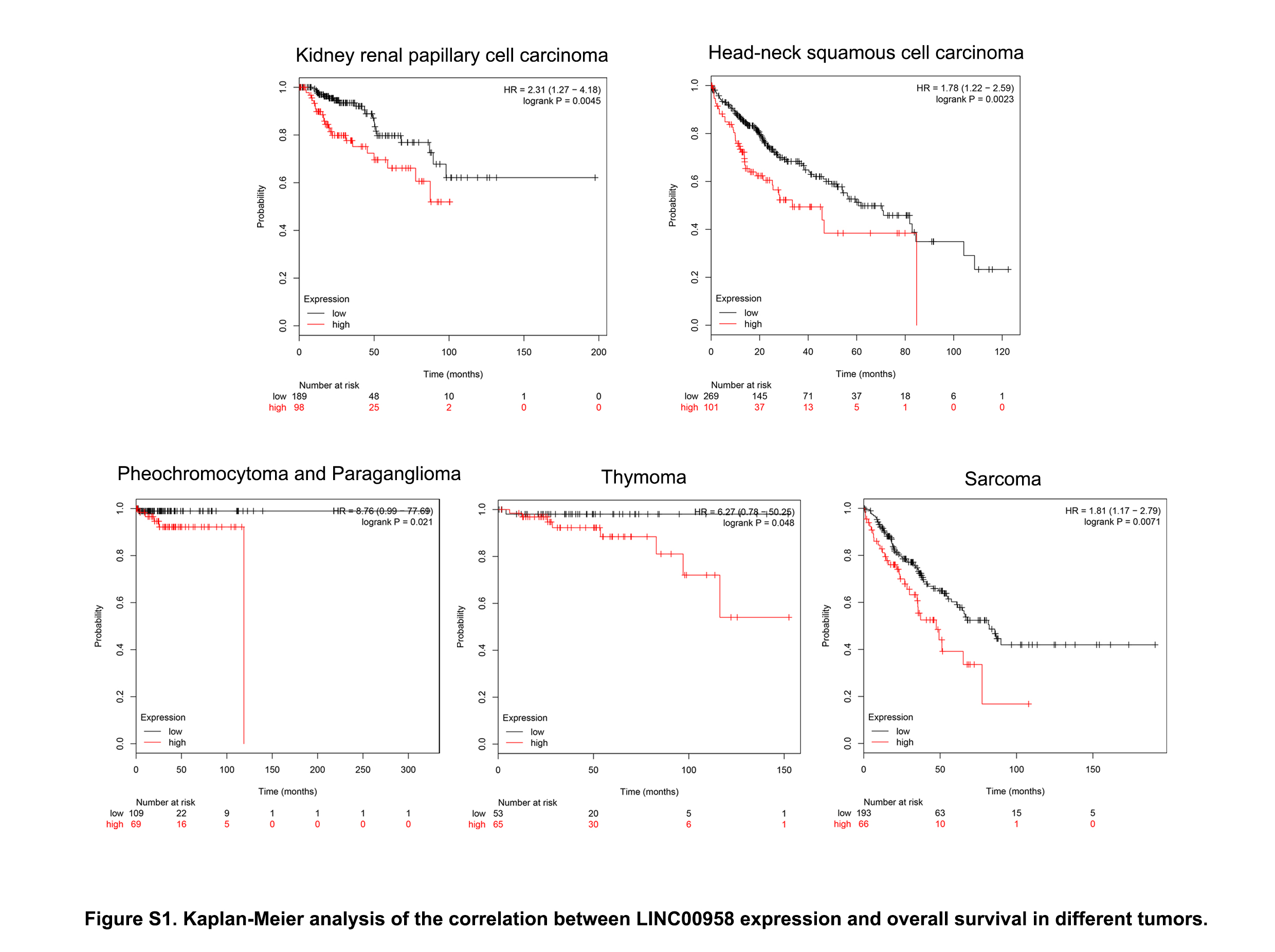
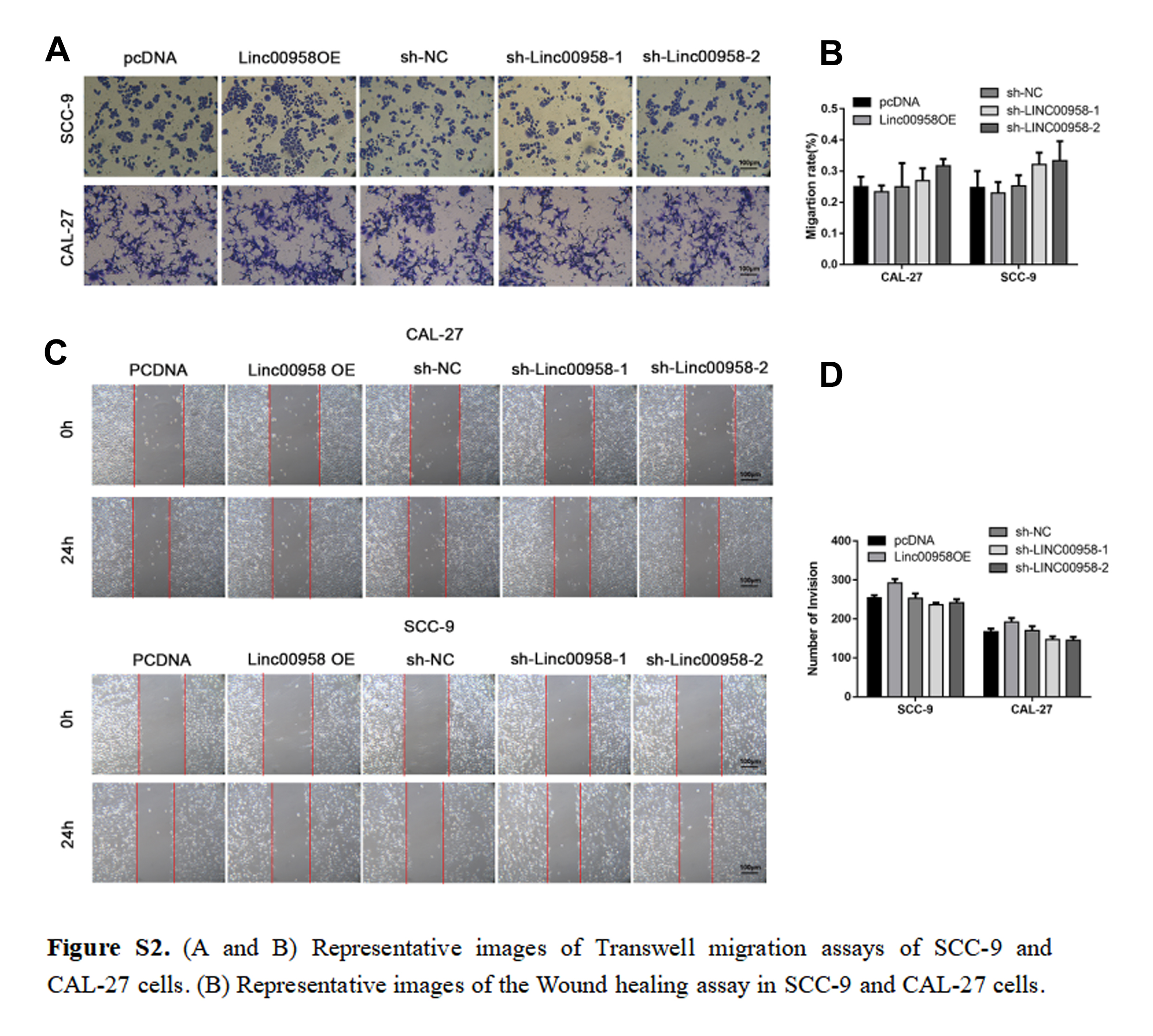
Data were expressed as mean ± SD of three independent experiments with GraphPad Prism softwareversion 6.0. Statistical analysis was performed using Statistical Package for Social Sciences (SPSS) software (version 16.0). Depending on the type of data, the appropriate statistical methods were used, including the t-test, one-way ANOVA, chi-square test, and linear correlation analysis. The Kaplan–Meier method with the log-rank test was used to compare the survival rate of TSCC patients based on LINC00958 expression. Survival data were evaluated using univariate and multivariate Cox proportional hazards models. p < 0.05 was considered statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}