The presence of LCRs is a trait of a broad spectrum of proteins, and appears to contribute to the antigenic variability in both viral and cellular pathogen populations. It is generally agreed that their origin is associated with errors due to polymerase slippage during genome replication and/or unequal crossover recombination events27,28. The processes that lead to their preservation in highly streamlined genomes, such as those of most RNA viruses, are not well understood, and their tempo and mode of evolution remain open issues. However, the extraordinary conservation of the two small LCRs reported here (Spike LCR-3, Spike LCR-4) in the rapidly-spreading Delta variant suggests that they are part of its hallmark traits. Accordingly, a detailed analysis of their frequency and phenotypic significance may contribute to the understanding of the origin of this variant’s increased transmissibility. A possible new variant of Delta, known as Delta Plus, was recently reported by the Public Health England service in a June 11, 2021 bulletin. This variant has a K417N mutation in the receptor-binding domain (RBD) of the spike protein30,31. Although the Delta Plus variant is considered a VOC, its properties are still being investigated. Our analysis of the spike protein of this putative variant shows that it has the same LCRs present in the SARS-CoV-2 Delta spike protein itself.
The Spike LCR-1 (FVFLVLLPLV) is a highly hydrophobic region that consists of helix-forming residues, including phenylalanine, valine and leucine, and it is the major component of the signal peptide (amino acids 1–13) located upstream of the N-terminus domain32 (Fig. 3). In the lumen of the endoplasmic reticulum this signal peptide plays a key role in guiding the spike protein to its membrane location by cellular signal peptidases33.
As noted above, the Kappa/Delta Spike LCR-3 and the Delta Spike LCR-4 regions are located in the spike S1 and S2 subunits, respectively. The mutation P681R detected in the Spike LCR-3 (SRRRARSVASQSIIA) (Fig. 3) at the furin cleavage site increases the polybasic nature of this region, which could augment its affinity with the furin protease34. In vitro experiments and SARS-CoV-2 infections in animal models have demonstrated that the P681R mutation enhances both the fusogenicity and pathogenicity of the virus35,36. The phylogenetic affinities between the Kappa- and Delta variants, both of which are part of the lineage B.1.61737,38 may explain in part the presence of these two mutations in the Delta- and the Kappa Spike LCR-3 (Figs. 1 and S1).
The SARS-CoV-2 spike protein is endowed with two heptad repeat motifs (HR1 and HR2) in its ectodomain that are involved in cell fusion, which is a key step in viral entry39,40. The Spike LCR-4 (LQNVVNQNAQALN) includes charged-neutral, polar (asparagine and glutamine) and hydrophobic amino acids (leucine, valine, and alanine), which are typical of heptad repeat motifs. The interaction of HR1 and HR2 leads to the formation of a six-helical bundle that mediates cell fusion39. Accordingly, it is possible that the asparagine (N) of the mutation D950N (Fig. 3) of the Spike LCR-4 may enhance the stabilization of the post-fusion hairpin conformation, since the conservation of the N and Q residues of HR1 is known to play an important role in the arrangement of hydrogen-bonding zippers that force HR2 to adopt its final conformation in SARS-CoV40. The structural relevance of this region has been demonstrated by studies with other RNA viruses, in which the use of fusion inhibitors that disrupt HR1-HR2 conformational changes, are known to limit viral entry41,42.
Although there may be minuscule variations in the LCRs length and/or amino acid composition, the segments described in this work fall well into the low complexity category, suggesting that their biased composition may confer adaptive advantages to the Delta variant. For instance, the polybasic Spike LCR-3, which includes several arginines in its N-terminus, is a highly conserved sequence located precisely in the furin cleavage site at spike S1/S2, which is essential for membrane fusion, and plays a key role in viral infection and transmission43,44.
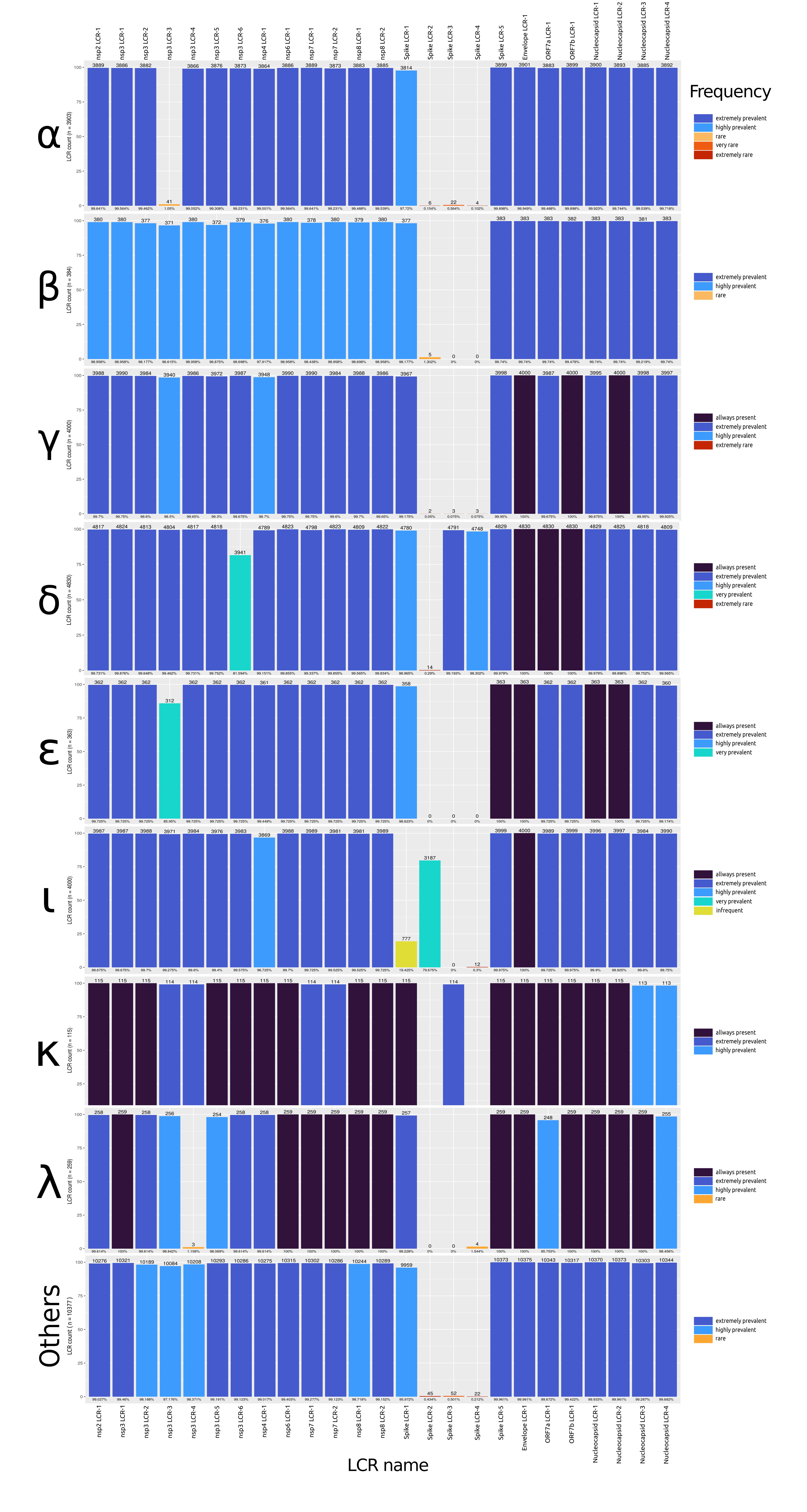
The use of the stringent cut-off value used here shows that, except for a limited number of sequences of the Spike LCR-3 and the Spike LCR-4 (39 and 82, respectively), these two LCRs are extremely prevalent (99.19% and 98.3% of all sequences) in our Delta variant sample (n = 4830). Although they display the biological traits of typical low complexity regions (Fig. 2), the multiple sequence alignments (Supplementary file 1 and 2) of the sequences that escape our cutoff values show single point mutations within these LCRs. These one-amino acid substitutions increase the complexity of the fragments, which prevents their detection by the methodology employed here. It is important to note that most of these sequences (34/39 and 82/82) lack some of the characteristic mutations (Supplementary file 1 and 2) that define the Delta variant29, including the prevalent P681R. It is thus possible they may be in fact sub-lineages of this VOC.
The SARS-CoV-2 Delta variant was detected in the late 202037, and the proteomics traits described here may contribute to explain in part its rapid worldwide expansion. The role of LCRs in enhancing sequence variability in surface proteins of viral and cellular pathogens has been postulated8,10. The extremely high conservation of the position and sequence of two LCRs (Spike LCR-3 and Spike LCR-4) in the Delta variant we have described is a strong indication of the importance of newly acquired raw material by slippage or recombination that may have undergone adaptative fine tuning, leading to the evolution and development of new functions or the improvement of existing ones.
LCRs can lead to variations in genome size in both viral and cellular systems45 and may have participated in some of the processes that led to an increase in the size of primitive cellular RNA genomes8,46. However, although compositionally biased sequences in SARS-CoV-2 are quite ubiquitous in most of the coronaviral proteins (Fig. 1 and S1), they do not contribute significantly to the increase of its genome size. In contrast, we hypothesize that the high conservation of the two LCRs in the Delta spike protein suggests that, together with the seven mutations present in this variant, they are part of the phenotypic traits associated with its high infectivity. Laboratory studies are required to confirm the possibility that the presence of compositionally biased segments in the Delta variant spike protein may be related to increased transmission, which is part of the defining features of VOCs and VOIs47–49.
{kind=link}
{kind=link}