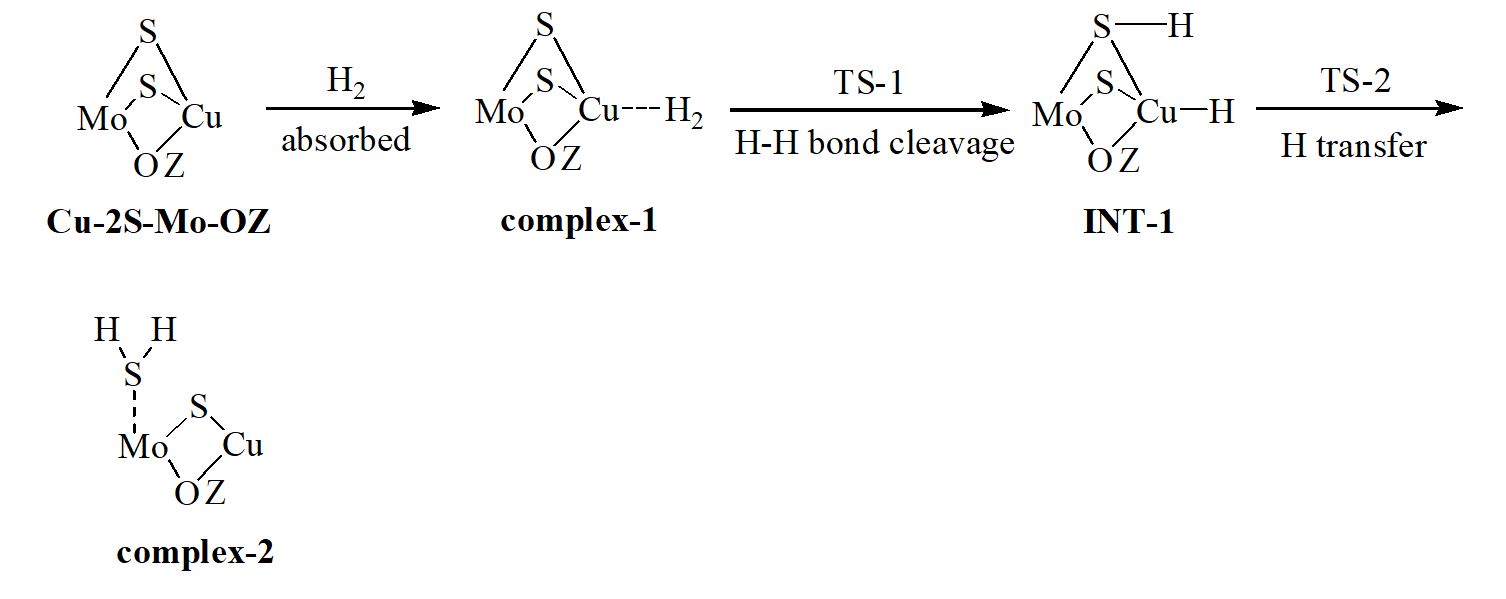
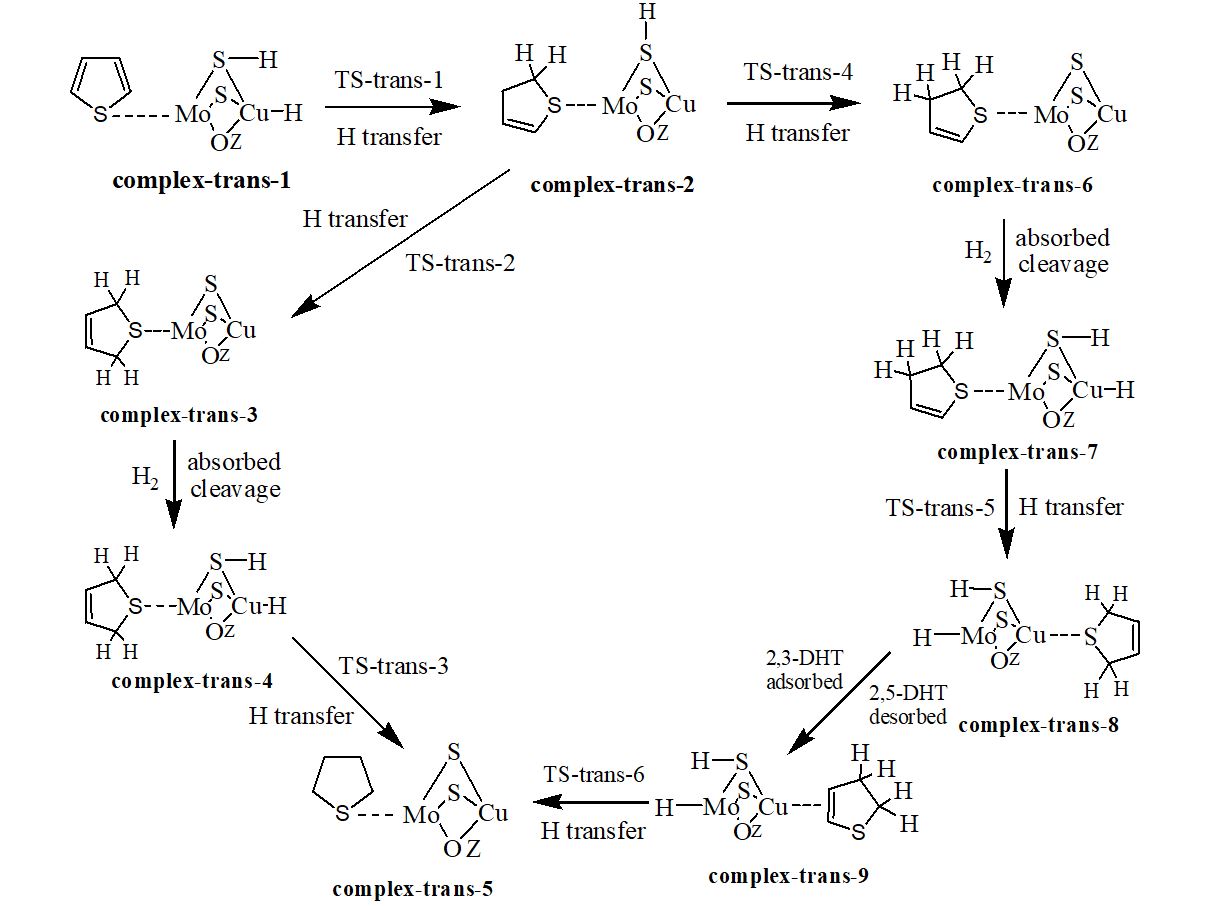
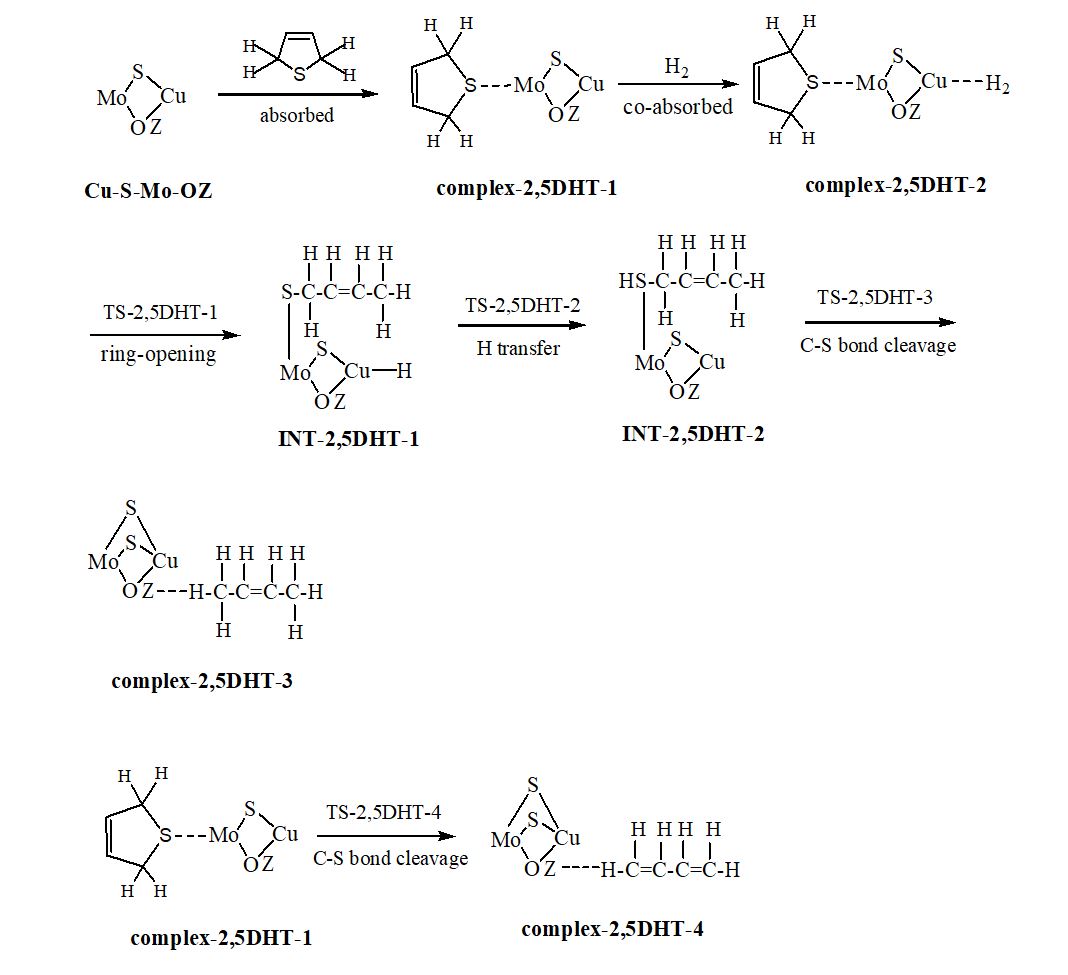
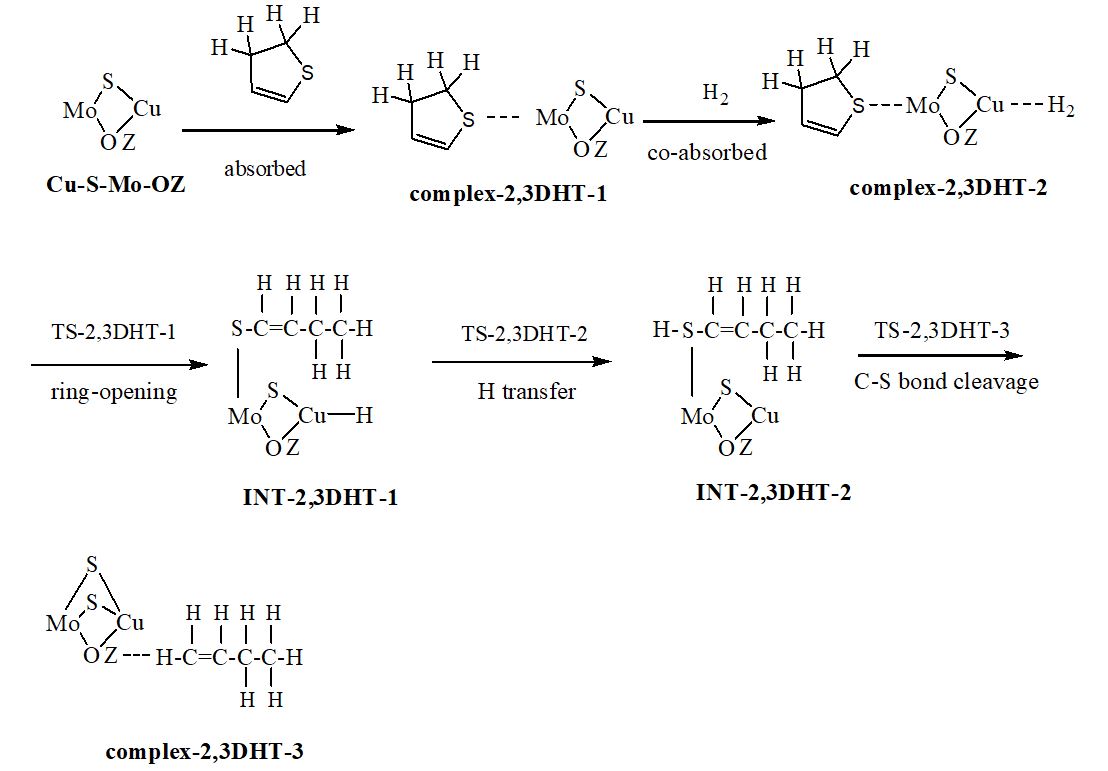
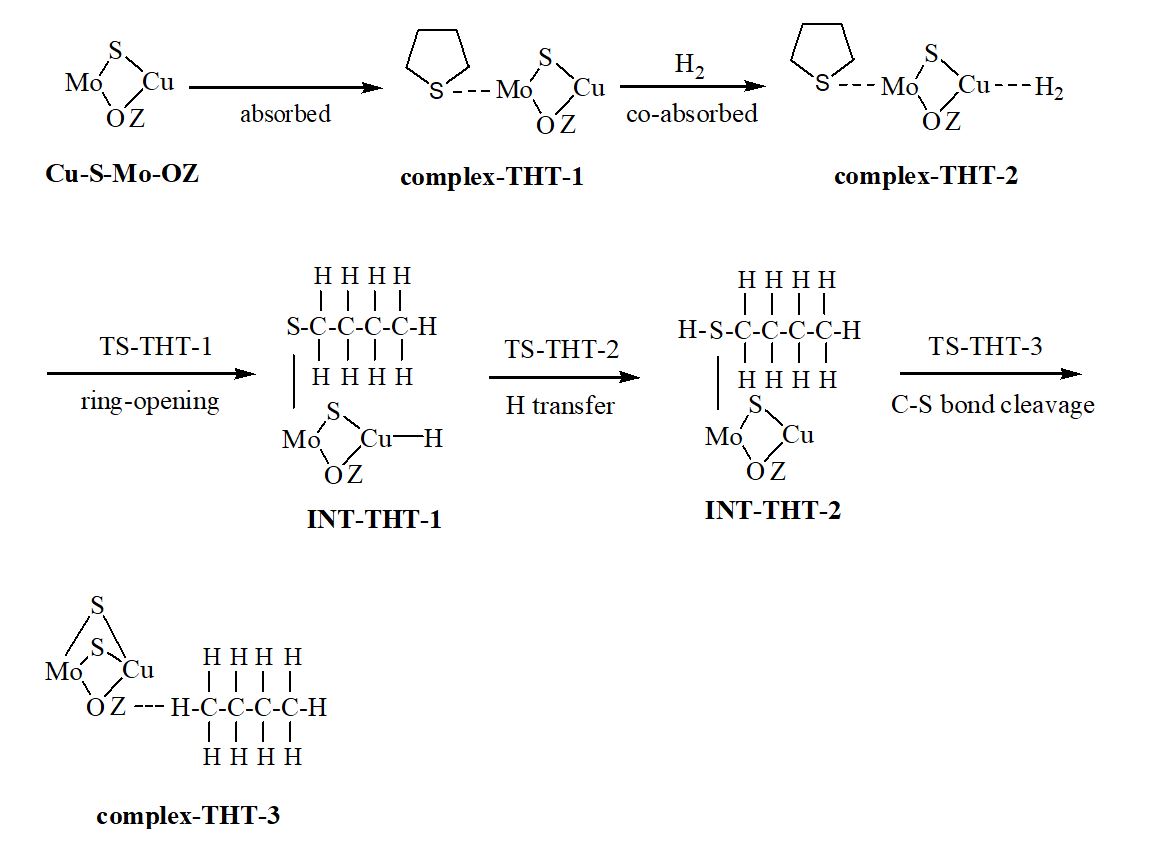
We have carried out a two-layer our own n-layered integrated molecular orbital and molecular mechanics (ONIOM) study on the hydrodesulfurization (HDS) mechanism of three hydrogenated thiophene derivatives over Cu-Mo modified FAU zeolite. The thiophene is hydrogenated relatively easily to 2,3-dihydrothiophene (2,3-DHT), 2,5-dihydrothiophene (2,5-DHT) and tetrahydrothiophene (THT) due to low free energy barriers. Hydrogenolysis desulfurization (HYD) and direct desulfurization (DDS) are discussed. Ring-opening, hydrogen transfer and C−S bond cleavage steps of thiophene derivatives are involved in the HYD process. The rate-determining steps are the hydrogen transfer step for 2,5-DHT and C−S bond cracking step for 2,3-DHT and THT. The concerted DDS pathway is probably more favorable than the HYD pathway in the desulfurization of 2,5-DHT. The hydrogenation of thiophene to 2,5-DHT, 2,3-DHT and THT and their HDS process are entropy-decreased; the formation of sulfur vacancy is entropy-increased. The difference charge density (DCD) analysis reveals that for the ring-opening process, the electrons are migrated from the organic chain to the Cu-Mo catalytic center. The reduced density gradient (RDG) plots indicate that both a steric hindrance and a weak van der Waals attractive interaction exist between organic fragment and catalytic center for all transition states (TSs). The localized orbital locator (LOL) maps for all TSs suggest that there are strong covalent interactions between the atoms in the forming chemical bonds and weak van der Waals interactions between the atoms in the breaking chemical bonds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}