A digital Gallen-KampMFB-595 instrument was used to measure melting points using open capillary tubes. The results were not corrected. KBr pellets were used to measure IR spectra using a Shimadzu FTIR-440 spectrometer. The MS-50 Kratos (A.E.I.) spectrometer, equipped with a data system, was used to obtain mass spectra at 70 eV. Chemical shifts in ppm units for 1H NMR (500 MHz) and 13CNMR (125 MHz) were measured using a Bruker model Ultra Shield NMR spectrumrometer in DMSO-d6 using tetramethyl silane (TMS) as an internal reference. Cairo University's Microanalytical Center conducted the elemental studies (percent C, H, and N). The appropriate precautions in handling moisture-sensitive compounds were taken. Solvents were dried by standard techniques. The monitoring of the progress of all reactions and homogeneity of the synthesized compounds were carried out and were run using thin-layer chromatography (TLC) aluminum sheets silica gel 60 F254 (Merck).
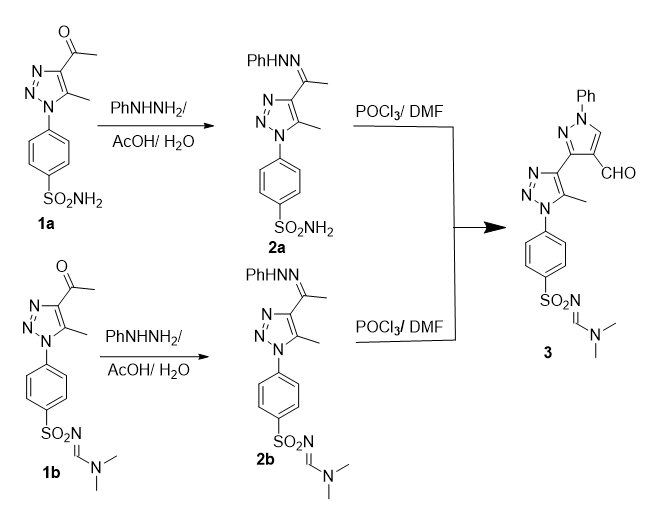
General procedure for synthesizing of hydrazones derivatives 2a and 2b: To 1a and 1b (10 mmol) in acetic acid (100 mL) and water (10 mL) phenylhydrazine (11 mmol) was added. The reaction mixture was stirred at room temperature until completion (TLC, 5 h). The solid which was formed was filtered off, washed with water, dried and crystallized from acetic acid to give:
4-[5-methyl-4-(1-(2-phenylhydrazineylidine) ethyl)-1H-1,2,3-triazol-1-yl]benzene-sulfomide (2a). Colorless crystals, yield 90%, m.p. = 256–258 oC. IR (KBr, cm− 1) ν = 3323, 3165 (NH2), 3136 (NH), 1371, 1141 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 9.35 (s, 1H, NH), 8.06 (d, 2H, J = 8.5 Hz, Ar-H), 7.88 (d, 2H, J = 8.5 Hz, Ar-H), 7.60 (s, 2H, NH2), 6.73–7.17( m, 5H, Ph-), 2.62 (s, 3H, CH3-), 2.43 (s, 3H, CH3-). Anal. Calcd. For. C17H18N6O2S (370.43): C, 55.12; H, 4.90; N, 22.69. Found: C, 55.20; H, 4.80; N, 22.80.
N,N,-dimethyl-N’-[(4-(5-methyl-4-(1-(2-phenylhydrazineylidene)ethyl)-1H-1,2,3-triazol-1yl)sulfonyl]formimidamide (2b). Colorless crystals, yield 88%, m.p. = 179–180 oC. IR (KBr, cm− 1) ν = 3321, (NH), 1629 (N = CH-), 1373, 1149 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 9.33 (s, 1H, NH), 8.29 (s, 1H, N = CH), 8.02 (d, 2H, J = 8.5 Hz, Ar-H), 7.82 (d, 2H, J = 8.5 Hz, Ar-H), 6.73–7.17( m, 5H, Ph-), 3.16 (s, 3H, N-CH3), 2.94 (s, 3H, N-CH3) 2.63(s, 3H, CH3-), 2.46(s, 3H, CH3-). MS, m/z (%): 425 [M+] (100). Anal. Calcd. For. C20H23N7O2S (425.51): C, 56.45; H, 5.45; N, 23.04. Found: C, 56.30; H, 5.60; N, 23.10.
N’-[(4-(4-(4-formyl-1-phenyl-1H-pyrazol-3-yl)-5-methyl-1H-1,2,3-triazol-1-yl)phenyl)sulfonyl]-N,N-dimethylformimidamide (3). Phosphorous oxychloride (25 mmol) was added to DMF (100 mL) at 0 oC and stirred for 30 min. 2a or 2b (10 mmol) were added slowly to this mixture and stirred for 5 h. The crude reaction was then quenched into water (1 L) and stirred for an additional 1 h the solid formed was separated, washed with water, dried, and crystallized from methanol to afford 3.
Colorless crystals, yield 88%, m.p. = 250–251 oC. IR (KBr, cm− 1) ν = 1672, (CO), 1633 (N = CH-), 1342, 1153 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 10.52 ( s, 1H, CHO), 9.28 (s, 1H, pyrazole H-5), 8.02 (s, 1H, N = CH), 8.00 (d, 2H, J = 8.6 Hz, Ar-H), 7.82 (d, 2H, J = 8.6 Hz, Ar-H), 7.86–7.54( m, 5H, Ph-), 3.14 (s, 3H, NCH3), 2.92 (s, 3H, NCH3) 2.66 (s, 3H, CH3-). MS, m/z (%): 463 [M+] (100). Anal. Calcd. For. C22H21N7O3S (463.52): C, 57.01; H, 4.57; N, 21.15. Found: C, 57.20; H, 4.60; N, 21.10.
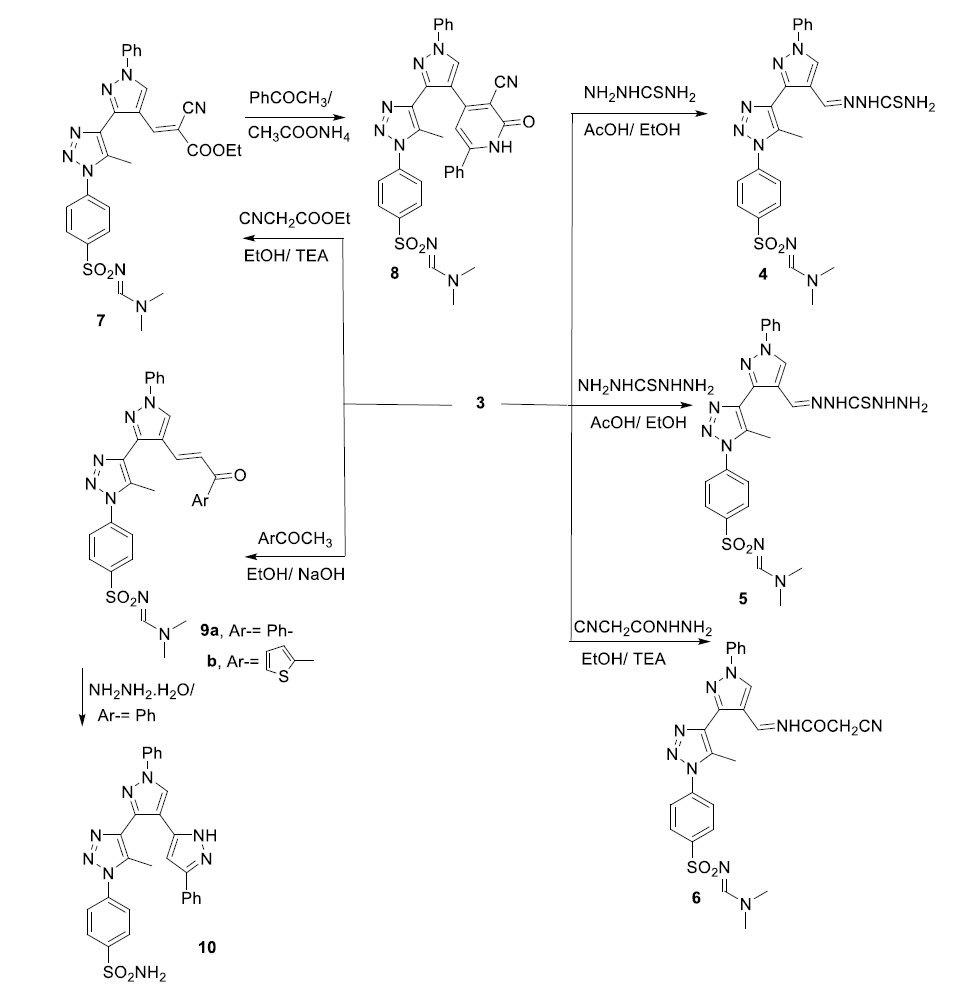
General procedure for the synthesis of compounds 4, 5, and 6. A solution of 3 (4.63 g, 10 mmol) and thiosemecarbazide or carbothidihydrazide or cyanacetic acid hydrazide (11 mmol ) in a mixture of acetic acid/ ethanol ( 1:1) was refluxed for 6 h. The solid formed on boiling was filtered of, dried and crystallized from acetic acid to afford:
2-[(3-(1-(4-(N-(dimethylamino) methylene) sulfamoyl) phenyl)-5-methyl-1H-1,2,3,-triazol-4-yl)-1-phenyl-1H-pyrazol-4-yl)methylene]hydrazine-1-carbothioamide (4). Pale yellow crystals in 86% yield; m.p. 234–236 oC. IR (KBr, cm− 1) ν = 3427( NH),3329, 3253 (NH2), 1637 (C = N), 1346, 1143 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 11.55 (s, 1H, NH), 9.29 (s, 1H, pyrazole H-5), 8.74 (s, 1H, CH = N), 8.30 (s,2H, NH2), 8.04 (d, 2H, J = 8.5 Hz, Ar-H), 7.91 (d, 2H, J = 8.5 Hz, Ar-H), 7.89 (s, 1H, N = CH), 7.59–7.37 ( m, 5H, Ph-H), 3.17 (s, 3H, NCH3), 2.94 (s, 3H, NCH3) 2.68 (s, 3H, CH3-). Anal. Calcd. For. C23H24N10O2S2 (536.63): C, 51.58; H, 4.51; N, 26.10. Found: C, 51.60; H, 4.70; N, 26.20.
N’-[(4-(4-(4-(2-(hydrazinecarbothioyl)hydrazineylidene)methyl)-1-phenyl-1H-pyrazol-3yl)-1H- -5-methyl-1H-1,2,3-triazol-1-yl) phenyl)sulfonyl]-N,N,-dimethylformimidamine (5). Pale yellow crystals in 86% yield; m.p. 202–204 oC. IR (KBr, cm− 1) ν = 3232 − 3138( 2NH, NH2)), 1633 (C = N), 1338, 1147 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 11.92 (s., 1H, NH), 11.77 (s, 2H, NH2), 9.92 (s, 1H, NH), 8.79 (s, 1H, pyrazole H-5), 8.29 (s, 1H, CH = N), 8.04 (d, 2H, J = 8.5 Hz, Ar-H), 7.90 (d, 2H, J = 8.5 Hz, Ar-H), 7.87 (s, 1H, N = CH), 7.58–7.40 ( m, 5H, Ph-H), 3.17 (s, 3H, NCH3), 2.94 (s, 3H, NCH3) 2.69 (s, 3H, CH3-). Anal. Calcd. For. C23H25N11O2S2 (551.65): C, 50.08; H, 4.57; N, 27.93. Found: C, 50.20; H, 4.70; N, 27.80.
N’-[(4-(4-(4-(2-(2-cyanoacetyl) hydrazineylidene)methyl)-1-phenyl-1H-pyrazol-3-yl)-1H- -5-methyl-1H-1,2,3-triazol-1-yl) phenyl)sulfonyl]-N,N,-dimethylformimidamine (6). Colorless crystals in 79% yield; m.p. 152–154 oC. IR (KBr, cm− 1) ν = 3217( NH), 2250(CN), 1697 (CO), 1627(C = N) (CO), 1344, 1149 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 11.81 (s., 1H, NH), 9.10 (s, 1H, pyrazole H-5), 8.74 (s, 1H, CH = N), 8.29 (s, 1H, N = CH), 8.02 (d, 2H, J = 8.6 Hz, Ar-H), 7.89 (d, 2H, J = 8.6 Hz, Ar-H), 7.58–7.37 ( m, 5H, Ph-H), 4.25 (s, 2H, CH2), 3.17 (s, 3H, NCH3), 2.94 (s, 3H, NCH3) 2.69 (s, 3H, CH3-). Anal. Calcd. For. C25H24N9O3S (530.17): C, 56.59; H, 4.56; N, 23.76. Found: C, 56.30; H, 4.50; N, 23.60.
Ethyl-2-cyano-3-[3-(1-(4-(N-(-dimethylamino) methylene) sulfamoyl) phenyl)-5-methyl-1H-1,2,3-triazole-4-yl] acrylate (7). A solution of 3 (10 mmol), ethyl cyanoacetate (1.13 mL, 10 mmol) in the presence of 3 drops of triethylamine in absolute ethanol (20 mL) was refluxed for 5 h. The solution was cooled and poured onto crushed ice. The resulting solid was filtered, dried and recrystallized from ethanol to yield 7. Yellow crystals in 90% yield; m.p. 230–231 oC; IR (KBr, cm− 1) ν = 2220 (CN), 1703 (COOEt), 1624 (C = C), 1336, 1151 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 9.21 (s, 1H, pyrazole H-5), 8.17 (s, 1H, N = CH), 8.00 (s, 1H, CH = C), 7.89 (d, 2H, J = 8.6 Hz, Ar-H), 7.86 (d, 2H, J = 8.6 Hz, Ar-H), 7.56–7.45( m, 5H, Ph-), 4.29 (q, 2H, J = 7.6 Hz, CH2, ester), 3.13 (s, 3H, NCH3), 2.92 (s, 3H, NCH3) 2.66(s, 3H, CH3-), 1.27 (t, 3H, J = 7.65 Hz, CH3-), MS, m/z (%): 558 [M+] (100). Anal. Calcd. For. C27H26N8O4S (558.62): C, 58.05; H, 4.69; N, 20.06. Found: C, 58.20; H, 4.80; N, 20.10.
N’-[(4-(4-(4-(3-cyano-2-oxo-6-phenyl-1,2-dihydropyridin-4-yl)-1-phenyl-1H-pyrazol-3-yl)-5-methyl-1H-1,2,3-triazol-1-yl)phenyl)sulfonyl]-N,N-dimethylformimidamide (8). A mixture of 4 (2.79 g, 5 mmol), ammonium acetate (2.73 g, 10 mmol) a acetophenone (5 mmol) in absolute ethanol (50 mL) was refluxed for 12 h. The solid which was precipitated during heating, was filtered, washed with water, dried, and crystallized from acetic acid to afford 8 as pale yellow crystals in 80% yield; m.p. 264–266 oC. IR (KBr, cm− 1) ν = 3360( NH), 2218 (CN), 1655 (CO), 1337, 1154 (SO2); 1H NMR (500 MHz, δ, pmm, DMSO-d6): δ = 12.54 ( brs., 1H, NH), 9.28 (s, 1H, pyrazole H-5), 8.12 (s, 1H, N = CH) ,8.00 (d, 2H, J = 8.6 Hz, Ar-H), 7.90 (d, 2H, J = 8.6 Hz, Ar-H), 7.74 (s, 1H, pyridine H-5), 7.56–7.30 ( m, 10H, 2 Ph-H), 3.15 (s, 3H, NCH3), 2.94 (s, 3H, NCH3) 2.66(s, 3H, CH3-). Anal. Calcd. For. C33H27N9O3S (629.70): C, 62.94; H, 4.32; N, 20.02. Found: C, 62.80; H, 4.50; N, 20.10.
General procedure for the synthesis of chalcones 9a-c: To a well-stirred solution of acetophenone or 2-acetylthiophene or 2-acetylpyridine 3 (10 mmol) in alcoholic NaOH (5 %, 25 ml) at 0–5 oC a solution of 3 (2.32 g, 5 mmol) was added gradually. Stirring was continued for 10 h at r.t. the resulting precipitate was filtrated, washed with water, dried, and crystallized from acetic acid to give.
N,N-dimethyl-N’-[(4-(5-methyl-4-(4-(3-oxo-3-phenylprop-1-en-1-yl)-1-phenyl-1H-pyrazol-3-yl)-1H-1,2,3-triazol-1-yl)phenyl)sulfonyl]formimidamide (9a). Yellow crystals in 86% yield; m.p. 190-191oC. IR (KBr, cm− 1) ν = 1678 (CO), 1631 (C = N), 1597 (C = C), 1340, 1163 (SO2); 1H NMR (DMSO-d6): δ = 9.49 (s, 1H, pyrazole H-5), 8.60 (s, 1H, CH = N), 8.10 (d, 2H, J = 8.5 Hz, Ar-H), 7.92 (d, 2H, J = 8.5 Hz, Ar-H), 7.80 (d, 1H, J = 15.30 Hz, CH = CH), 7.70 (d, 1H, J = 15.30 Hz, CH = CH), 7.59–7.45(m, 10H, 2Ph-H), 3.13 (s, 3H, NCH3), 2.94 (s, 3H, NCH3) 2.67 (s, 3H, CH3-). Anal. Calcd. For. C30H27N7O3S (565.65): C, 63.70; H, 4.81; N, 17.33. Found: C, 63.80; H, 4.70; N, 17.50.
N,N-dimethyl-N’-[(4-(5-methyl-4-(4-(3-oxo-3-(thiophen − 2-yl) prop-1-en-1-yl)-1-phenyl-1H-pyrazol-3-yl)-1H-1,2,3-triazol-1-yl)phenyl)sulfonyl]formimidamide (9b). Yellow crystals in 80% yield; m.p. 166–168 oC. IR (KBr, cm− 1) ν = 1643 (CO), 1631 (C = N), 1597 (C = C), 1342, 1159 (SO2); 1H NMR (DMSO-d6): δ = 9.49 (s, 1H, pyrazole H-5), 8.60 (s, 1H, CH = N), 8.48( d, 1H, J = 16.70 Hz, CH = CH), 7.94 (d, 2H, J = 8.5 Hz, Ar-H), 7.92 (d, 2H, J = 8.5 Hz, Ar-H), 7.59–7.45(m, 9H, CH = CH, Ph-H, 3 thiophene -H)), 3.16 (s, 3H, NCH3), 2.90 (s, 3H, NCH3) 2.66 (s, 3H, CH3-), MS, m/z (%): 571 [M+] (43). Anal. Calcd. For. C28H25N7O3S2 (571.67): C, 58.83; H, 4.41; N, 17.15. Found: C, 58.70; H, 4.60; N, 17.20.
4-(4-(1',5-diphenyl-1'H,2H-[3,4'-bipyrazol]-3'-yl)-5-methyl-1H-1,2,3-triazol-1-yl)benzenesulfonamide (10). To a solution of appropriate chalcones 9a (2.78g, 5 mmol) in ethanol (30 ml), hydrazine hydrate 80% (12 mmol) was added. The reaction mixture was refluxed for 6 h. Left to cool to room temperature. The formed solid product was filtered, dried, and recrystallized from EtOH to afford 10 as colorless crystals in 82% yield; m.p. 180–182 oC. IR (KBr, cm− 1) ν = 3218 (NH), 1631 (C = N), 1597 (C = C), 1340, 1163 (SO2); 1H NMR (DMSO-d6): δ = 11.21 (s, 1H, NH), 9.49 (s, 1H, pyrazole H-5), 8.60 (s, 1H, CH = N), 8.20(s, 1H, pyrazole- H), 7.94 (d, 2H, J = 8.5 Hz, Ar-H), 7.92 (d, 2H, J = 8.5 Hz, Ar-H), 7.59–7.45(m, 10H, 2Ph-H)), 3.19 (s, 3H, NCH3), 2.92 (s, 3H, NCH3) 2.65 (s, 3H, CH3-). Anal. Calcd. For. C27H22N8O2S (522.16): C, 62.06; H, 4.24; N, 21.44. Found: C, 62.20; H, 4.80; N, 21.70.
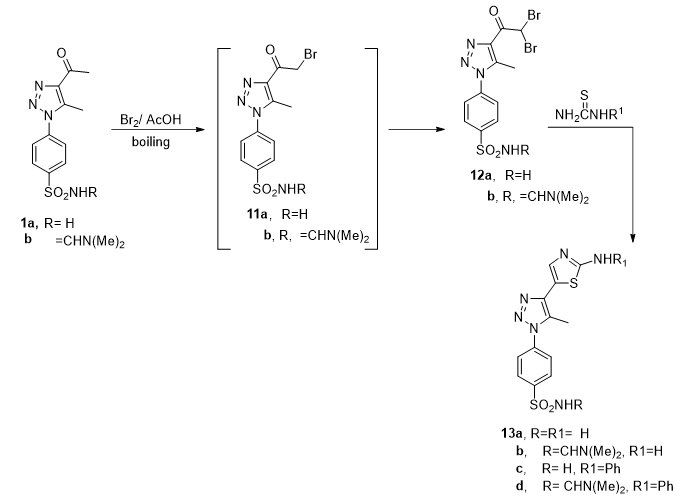
General procedure for the synthesis of compounds 11a, 12a, and 12b. To a stirred cold solution of compound 1a or 1b ( 10 mmol) in acetic acid, bromine (15 mmol) is add-drop wisely within 1h and the stirring is continued at room temperature for another 4 hrs. The reaction mixture was heated at 80oC for another 6 hrs. The solid which was formed on heating filtered off, washed with ethanol ( 10 ml), dried, and crystallized from acetic acid to give :
4-[4-(2-bromoacetyl)-5-methyl-1H-1,2,3-triazol-1-yl] benzenesulfonamide (11 a). Colorless crystals, m.p. 230-232oC. IR (KBr, cm− 1) ν = 3340, 3194 (NH2), 1681 (C = O), 1330, 1161 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 8.01 (d, 2H, J = 8.6 Hz, Ar–H), 7.90 (d, 2H, J = 8.6 Hz, Ar–H), 7.57 (s, 2H, NH2), 4.66 (s, 2H, CH2), 2.62 (s, 3H, CH3). Anal. Calcd for C11H11BrN4O3S (359.20): C, 36.78; H, 3.09; N, 15.60. Found C, 36.90; H, 3.20; N, 15.70.
4-[4-(2,2-dibromoacetyl)-5-methyl-1H-1,2,3-triazol-1-yl) benzene sulfonamide (12a). Colorless crystals in 90% yield; m.p. 222–224 oC. IR (KBr, cm− 1) ν = 3226, 3103 (NH2), 1705 (C = O), 1338, 1163 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 8.0 (d, 2H, J = 8.5 Hz, Ar–H), 7.92 (d, 2H, J = 8.5 Hz, Ar–H), 7.62 (s, 2H, NH2), 7.57 (s,1H, CH), 2.62 (s, 3H, CH3). 13C NMR (125 MHz, δ, pmm, DMSO-d6) δc 10.09 (CH3), 41.86 (CH), 126.32, 127.26, 137.02 (= CH triazole), 137.42 (= CH triazole), 141.58, 145.56,181.32 (C = O). Anal. Calcd for C11H10Br2N4O3S (438.09): C, 30.16; H, 2.30; N, 12.79. Found C, 30.10; H, 2.40; N, 12.90.
N-[[4-[4-(2,2-dibromoacetyl)-5-methyl-1H-1,2,3-triazol-1-yl] phenyl] sulfonyl]-N, N-dimethylformimidamide (12b). Colorless crystals in 92% yield; m.p. 214–216 oC. IR (KBr, cm− 1)ν = 2991 (CH), 1699(C = O), 1338, 1151 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 8.28 (s, 1H, CH = N), 8.02(d, 2H, J = 8.6 Hz, Ar–H), 7.86 (d, 2H, J = 8.6 Hz, Ar–H),, 7.57 (s, 1H, CH), 3.16(s, 3H, NCH3), 2.93(s, 3H, NCH3), 2.62 (s, 3H, CH3) .13C NMR (125 MHz, δ, pmm, DMSO-d6) δc 10.05 (CH3), 35.20 (NCH3), 41.04 (CH), 126.06, 127.50, 136.92 (= CH triazole), 137.35 (= CH triazole), 141.59, 144.60, 160.16 (N = C),181.31 (C = O). MS, m/z (%): 493 [M+](56), 445 [M+-N(CH3)2] (4). Anal. Calcd for C14H15Br2N5O3S (493.17): C, 34.10; H, 3.07; N, 14.20. Found C, 34.20; H, 3.20; N, 14.30.
General procedure for the synthesis of compounds 13a-d: A solution of 12a or 12b (10 mmol), and thioura or N-phenylurea in a mixture of acetic acid/ ethanol (50 ml, 1:1) was refluxed for 10h. The solid formed on boiling was filtered of, dried and crystallized from acetone to afford:
4-[4-(2-aminothiazol-5-yl)-5-methyl-1H-1,2,3-triazol-1-yl] benzene sulfonamide (13a). Pale yellow crystals, 83% yield m.p. 268–270 o C. IR (KBr, cm− 1) ν = 3329, 3228 (2NH2), 2889 (CH), 1624(C = N), 1334, 1155 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 8.66 (brs., 2H, NH2), 8.07 (d, 2H, J = 8.5 Hz, Ar–H), 7.90 (d, 2H, J = 8.5 Hz, Ar–H), 7.88 (s, 2H, NH2), 7.16 (s,1H, thiazole- H4), 2.52 (s,3H, CH3) .13C NMR (125 MHz, δ, pmm, DMSO-d6) δc = 9.99, 103.89, 125.82, 127.31, 130.11, 133.07, 135.47, 137.62, 145.20, 169.99. MS, m/z (%): 336 [M+] (22). Anal. Calcd for C12H12N6O2S (336.39): C, 42.85; H, 3.60; N, 24.98. Found C, 42.70; H, 3.50; N, 24.90.
N'-((4-(4-(2-aminothiazol-5-yl)-5-methyl-1H-1,2,3-triazol-1-yl)phenyl)sulfonyl)-N,N-dimethylformimidamide (13 b). Yellow crystals, 82% yield, m.p. 234–236 o C. IR (KBr, cm− 1) ν = 3354, 3263 (NH2), 2999 (CH), 1635(C = N), 1340, 1143 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 8.66 (brs, 2H, NH2), 8.28 (s, 1H, CH = N), 8.01 (d, 2H, J = 8.5 Hz, Ar–H), 7.82 (d, 2H, J = 8.5 Hz, Ar–H), 7.14 (s,1H, thiazole- H4), 3.16 (s, 3H,NCH3), 2.93 (s, 3H, NCH3), 2.52 (s, 3H, CH3). 13C NMR (125 MHz, δ, pmm, DMSO-d6) δc = 9.9, 103.9, 125.5 (2C), 125.8, 127.2, 127.3 (2C), 130.1, 132.9, 135.4, 137.6, 144.8, 145.2, 169.90. Anal. Calcd for C15H17N7O2S2 (391.47): C, 46.02; H, 4.38; N, 25.05. Found C, 46.10; H, 4.20; N, 25.20.
4-[5- methyl-4-(2-(phenylamino) thiazol-5-yl)- 1H-1,2,3-triazol-1-yl] benzene sulfonamide (13c). Yellow crystals 85% yield, m.p. 254 − 25 o C. IR (KBr, cm− 1) ν = 3329, 3242(NH2), 3147 (NH), 1618(C = N), 1338, 1166 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ 10.51 (s, 1H, NH), 8.07 (d, 2H, J = 8.5 Hz, Ar–H), 7.93 (d, 2H, J = 8.5 Hz, Ar–H), 7.65 (m, 3H, Ph-H), 7.29 (s,1H, thiazol –H4), 7.20 (s, 2H, NH2), 6.96 (m, 2H, Ph-H), 2.58 (s,3H, CH3) .13C NMR (125 MHz, DMSO-d6) δc = 10.1, 48.6, 92.4, 104.2, 117.3, 121.9, 125.6 (2C), 127.2 (2C), 129.0, 133.0, 138.00, 138.2, 139.1, 140.5, 144.2, 162.6. Anal. Calcd for C18H16N6O2S2 (412.49): C, 52.41; H, 3.91; N, 20.37. Found C, 52.60; H, 3.60; N, 20.50.
N,N- dimethyl amino-N'-[(4-(5-methyl-4-(2-(phenylamino)thiazol-5-yl)- 1H-1,2,3-triazol-1-yl)phenyl)sulfonyl]formimidamide (13 d). Pale yellow crystals, 78% yield, m.p. 265–267 o C. IR (KBr, cm− 1) ν = 3263 (NH), 1635(C = N), 1340, 1143 (SO2). 1H-NMR (500 MHz, δ, pmm, DMSO-d6): δ = 10.36 (s, 1H, NH), 8.28 (s, 1H, CH = N), 8.01 (d, 2H, J = 8.5 Hz, Ar–H), 7.84 (d, 2H, J = 8.5 Hz, Ar–H), 7.67 (s,1H, thiazole- H4), 7.28–6.93 ( m, 5H, Ph-H), 3.16 (s, 3H,NCH3), 2.93 (s, 3H, NCH3), 2.69 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δc = 10.1, 35.2, 41.2, 92.37, 104.2, 117.0, 125.6 (2C), 125.7 (2C), 127.5 (2C), 129.0, 129.1, 139.82, 140.5, 141.1, 142.2, 160.2, 162.6, 163.8. Anal. Calcd for C21H21N7O2S (467.57): C, 53.95; H, 4.53; N, 20.97. Found C, 53.80; H, 4.70; N, 20.80.
X-ray crystallography of compound 3.
A single crystal of compound 3 was obtained by slow evaporation at room temperature, from dimethylformamide (DMF). The crystal structure was solved and refined using MaXus (Bruker Nonius, Deflt and Mac Science, Japan)24. Mo Kα radiation (λ = 0.71073 Ǻ) and a graphite monochromator were used for data collection. The chemical formula and ring labeling system are shown in Fig. 2. Crystal data for compound 3: C22H21N7O3S, Mr = 463.520; system, Monoclinic; space group, P21/c; a = 11.3957 (3)Å; b = 13.1843 (5)Å; c = 5.1206 (5)Å; α = 90.00°; β = 106.413 (3)°; γ = 90.00°; V = 2179.21 (12)Å3; Z = 4; Dx = 1.413 Mg m− 3; range for data θ = 2.910-27.485 °; (Mo-Kα) µ = 0.190 mm− 1; T = 298 K; 16702 measured reflections; 4921 independent reflections; 2336 observed reflections; R(all) = 0.1595; R(gt) = 0.0623; wR(ref) = 0.2021; wR(gt) = 0.1604; S(ref) = 0.995; 4921 reflections; δ/σmax = 0.004; δρmax = 0.278eÅ3; δρmin = -0.346eÅ3. Crystallographic data for the structure 5 have been deposited with the Cambridge Crystallographic Data Center (CCDC) under the number 1574927. Copies of the data can be obtained, free of charge, on application to CCDC 12 Union Road, Cambridge CB2 1EZ, UK [Fax: +44-1223- 336033; e-mail: [email protected] or at www.ccdc.cam.ac.uk.
Molecular docking studies
Materials and methods
Three-dimensional structure of human cycline-dependantkinase CDK2, PDB:2FVD, was obtained from protein data bank. Energy minimization of newly designed compounds was done by employing discovery studio 2.5 for structure refinement. The geometry of all designed analogues is typed with CHARMm25 force field; then partial charges are calculated by Momany Rone method26. Further, they are optimized through a smart minimizer algorithm, which performs 1000 steps of steepest descent with a root mean-square (RMS) gradient tolerance of 0.1. Same as the preparation of ligands for the target, its active site was also passed with the energy minimization process and it was done using charm force field which is defined by the equation given below:
where E = total energy, Eb = bond potential energy, Eq and Ef = bond angle potential energy, Ew = torsion energy, Evdw = van der Waals interaction energy, Eel = electrostatic potential energy, Ehb = hydrogen bond energy, Ecr = energy constraints, and Ecj = energy function25.
CDOCKER (CHARMm-based DOCKER), a docking program provided by discovery studio 2.5, uses a CHARMm based molecular dynamics (MD) scheme to dock ligands into a receptor binding site, and then random conformations will be produced using high-temperature molecular dynamics. When these conformations are translated to the active site, candidate poses are then generated using random rigid body rotations followed by simulated annealing. CDOCKER offers all the advantages of full ligand flexibility (including bonds, angles, and dihedrals) and reasonable computation times. CDOCKER uses soft core potentials, which are found to be effective in exploring the conformational space of macromolecules used in various docking studies. The nonbonded interactions which involve van der Waals (vdW) and electrostatics are softened at different levels, except during the final minimization step27. Initially, ten conformations for each inhibitor are generated in the active site of the target enzyme, which is created as a spherical region with a diameter of 10 Ǻ. Simulated annealing is performed using a flexible ligand and a rigid protein. Receptor–ligand interactions are calculated from grid extension 8.0, random conformations are generated using specific molecular dynamics steps, and the system is heated to 700 K in 2000 steps, cooling steps to 5000, and cooling temperature to 300 K. The final refinement step of minimization is performed using full potential. Minimized docking poses are then clustered, based on a heavy atom RMSD approach. The ranking is based on the total docking energy, which is composed of the ligand’s intramolecular energy and the ligand-receptor interaction.
{kind=link}
{kind=link}
{kind=link}