Materials and reagents
IR780 was obtained from Sigma-Aldrich and used as received. TPZ was obtained from MedChemExpress and used as received. All other chemicals were purchased from Chongqing Chuandong Chemical (group) CO., Ltd. and used without further purification. All chemicals were analytical grade and used without further purification if not indicated otherwise. Dulbecco’s modified eagle’s medium (DMEM) was purchased from GE Healthcare Life Sciences HyClone Laboratories (HyClone, USA). Fetal bovine serum (FBS) and Trypsin EDTA Solution A were purchased from Biological Industries (BI, Israel). Cell Counting kit-8 (CCK-8) was purchased from Fdbio science CO., Ltd. (Hangzhou, China) and used as received. Dialysis tubes were purchased from Shanghai Lvniao Technology Group. Clear polystyrene tissue culture treated 6-well and 96-well plates were purchased from JET BIOFIL CO., Ltd. (Guangzhou, China). The deionized water was used in the experiment, which was purified from Milli-Q (Millipore, 18.2 MΩ cm-1).
Cells and animals
The human squamous carcinoma cells WSU-HN6, Murine macrophage-like cells RAW 264.7, human umbilical vein endothelial cells (HUVEC) and human cervical cancer cells (HeLa) were cultured as previously described [41, 42]. All specific pathogen free (SPF) mice were purchased from the Hunan SJA Laboratory Animal CO., LTD (Hunan, China) and housed under pathogen-free environment. All animal protocols were reviewed and approved by the Ethics Committee of the College of Stomatology, Chongqing Medical University (Approval No. 2021025). For all the experiments on animals in this research, mice were randomly (random number table method) allocated into each group. Grouping was done according to the experimental designs.
Cell membrane fragments fusion study
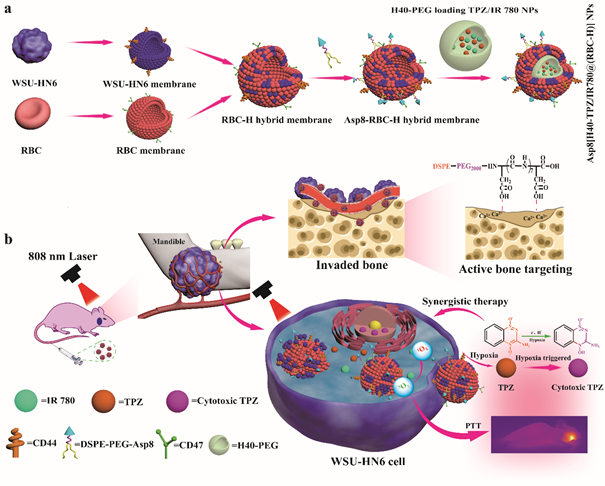
Before fusion, WSU-HN6 and RBC membrane fragments were prepared following the previously reported methods (See supplementary methods in Additional file 1). FÖrster resonance energy transfer (FRET) was employed to evaluate the erythrocyte and cancer cell membrane fusion process. Briefly, WSU-HN6 membranes was stained with DiI (detected at an excitation of 549 nm and emission of 565 nm) and DiO (Ex. 484 nm and Em. 501 nm). According to the theory that the weight of cell membrane was the double of membrane protein weight, erythrocyte membrane was added to WSU-HN6 membrane at erythrocyte membrane to WSU-HN6 membrane protein weight rations of 0:1, 1:1, 3:1,and 5:1, respectively [29]. Each group was sonicated for 1 min in an ice bath. After sonication, Samples were extruded through 1.0 × 103 nm, 400 nm, and 200 nm polycarbonate porous membrane to promote fusion of the two kinds of cell membrane. Finally, RBC-H hybrid membrane vesicles were obtained by centrifugation at 14000 × g for 0.5 h at 4 ℃ and resuspended in DEPC solution. Fluorescence spectrum for each group was recorded from 500 to 750 nm with an excitation wavelength at 484 nm. Fluorescence recovery of the donor (DiI) was used to monitor the changes in the fusion process.
Then, CLSM was employed to evaluate membrane colocalization. Briefly, DiI and DiO were first dissolved in dimethyl sulfoxide (DMSO). Then, 10 μL of DiO solution was added to 100 μL of the WSU-HN6 membrane solution. The mixed sample was stirred for 2 h away from light in an ice bath. Similarly, 10 μL of the DiI solution was added to 100 μL of RBC membrane solution. The sample was also stirred for 2 h away from light in an ice bath. After thorough mixture, dye labeled cell membrane solutions were centrifuged at 14000 × g for 0.5 h at 4℃ and washed four times using PBS to remove free DiO and the DiD dye. To fuse the two types of cell membranes, DiO-labeled WSU-HN6 membrane and DiI-labeled erythrocyte membrane were mixed in an ice bathe and sonicated for 2 min. After sonication, the mixed solution was extruded though a 1 μm, 400 nm, 200 nm polycarbonate porous membrane to prepare hybrid membrane fusion. Finally, the RBC-H hybrid membrane vesicles were obtained by centrifuged at 14000 × g for 0.5 h at 4 ℃ and resuspended in DEPC water. The RBC-M hybrid membrane (10 μL) was placed on a glass slide, naturally evaporated, and subjected to CLSM. DiI was excited using a 548 nm laser and the red mission collected at 565 nm. A physical mixture of DiO-ladeled WSU-HN6 membrane and DiO-labeled erythrocyte membrane without extrusion was employed as the control. To further validate the successfully fusion of both cell membranes, the immunogold staining assay was performed (See supplementary methods in Additional file 1).
Preparation and characterization of biomimetic NPs
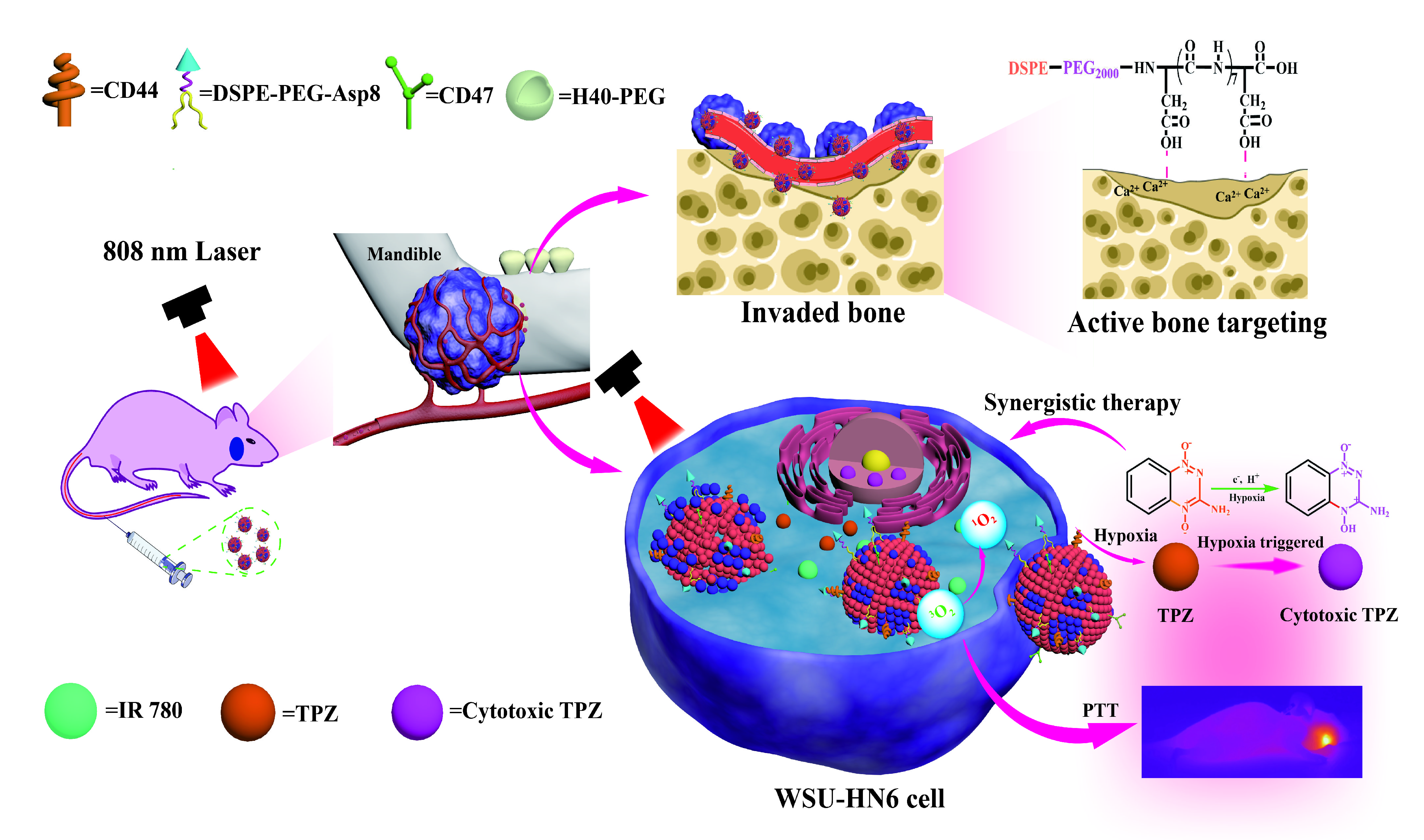
Synthesis of polyester H40-poly(ethylene glycol) (H40-PEG) and H40-PEG NPs were performed as previously described [43]. H40-PEG loading TPZ/IR780 nanoparticles were prepared using the oil-in-water emulsion solvent diffusion method as follows: H40-PEG (25 mg) and IR780 (2.5 mg) were dissolved in 5 mL of chloroform while TPZ was dissolved in 5 mL of deionized water [44]. Both solutions were mixed and ultrasonicated. After ultrasonication for 1 h in an ice bath, H40-TPZ/IR780 NPs were successfully synthesized. Brown precipitates were collected by centrifugation. Next, NPs were washed three times using deionized water for further analysis. The whole reaction was performed away from light.
RBC cell and WSU-HN6 cell membranes were mixed at a membrane weight ratio of 1:1. The mixture was sonicated at power level of 60 W for 5 min with a sequence of 1 min of sonication and 2 min of break in an ice bath. The RBC-H fused hybrid membrane was coated onto H40-PEG NPs or H40-TPZ/IR780 NPs through the extrusion method. The sonicated RBC-H hybrid membrane solution was added to H40-PEG NPs or H40-TPZ/IR780 NPs solution, and the mixture was successively extruded through 1 μm, 400 nm and 200 nm polycarbonate porous membrane to form hybrid membrane-coated NPs. Finally, biomimetic NPs were obtained by centrifugation (10000 rpm, 5min, 4 ℃), washed three times with PBS, and resuspended in a buffer solution for further characterization. To assess the stability of Asp8[H40-PEG@(RBC-H)] NPs, the changes in nanoparticle size were observed through DLS. Briefly, Asp8[H40-PEG@(RBC-H)] NPs was incubated with PBS, and the size changes in size over 72 h were measured. The H40-PEG NPs were used as a control.
Characterization of membrane protein
To evaluate membrane protein profiles of RBCs, WSU-HN6, H40-PEG@(RBC-H) NPs and Asp8[H40-PEG@(RBC-H)] NPs, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) assay was performed. Briefly, the radio immunoprecipitation assay (RIPA) buffer (Beyotime, China) was used to separate the surface proteins from RBC cells, WSU-HN6 cells, and hybrid membrane (RBC-H). Membrane protein samples were mixed with SDS-PAGE sample loading buffer (5 ×, Beyotime, China) and heated for 10 min at 100 ℃. Then, protein samples (at 20 μg mL-1 each) were loaded on 8% SDS-PAGE gels (Beyotime, China). After running at 80 V for 30 min, voltage was increased to 120 V and samples kept running for about 90 min. Coomassie brilliant blue was used to stain the resulting gels for 2 h. Stained gels were washed for 12 h, and the decolorizing solution was changed thrice during washing. Treated gels were captured by ChemiDOCTMXRS+System (Bio-Rad, USA).
To further characterize specific protein expression of cell membranes, resultant gels were transferred to polyvinylidene difluoride (PVDF) membranes (0.45 μm,Servicebio, China) for Western blot analysis. After blocking with 5% skimmed milk, PVDF membranes were probed with anti-CD47 (66304-1-Ig, Proteintech, Wuhan, China) and anti-CD44 (60224-1-Ig, Proteintech, Wuhan, China) overnight at 4 ℃. The PVDF membranes was incubated with horseradish peroxidase conjugated affinipure goat anti-mouse IgG (H+L) (SA00001-1, Proteintech, Wuhan, China) secondary antibody at room temperature for 1.5 h and washed three times (5 min time-1). Finally, an enhanced chemiluminescence detection kit (Beyotime, China) was used to visualize PVDF membranes. Signals of both special proteins were recorded on the ChemiDOCTMXRS+System (Bio-Rad, USA).
Hydroxyapatite binding assay
To investigate bone-targeted abilities of the biomimetic NPs, hydroxyapatite (HA) binding assay was performed in vitro as previously reported [45]. HA beads were suspended in PBS at 20 mg mL-1. Then, 600 μL of different DiI-loaded nanoparticles (DiI as the fluorescent probe with a concentration of 10 Μm for each sample) were mixed with 300 μL of HA suspension or 300 μL of the PBS as a control, followed by gentle shaking at 100 rpm for 1 h at 37 ℃. After centrifugation at 10,000×g for 5 min, the HA precipitate was separated from unbound nanoparticles in the supernatant. The DiI fluorescence of supernatant was quantified through a multi-functional microporous plate reader (SpectraMAX iD5, USA) at a wavelength of 549 nm. The degree of HA binding was calculated according to the following: Equation (1): HA binding rate (%) = (A-B)/A. Where A is the fluorescence intensity of DiI in the control group and B is the fluorescence intensity of DiI in the experimental groups.
Specific targeting WSU-HN6 cell line in vitro
HeLa cells, HUVEC cells and WSU-HN6 cells were seeded into 6-well plates with cell climbing slice at a density of 5.0 × 104 cells well-1 in 1.5 mL of complete DMEM, respectively. After incubation for 12 h, medium was replaced with fresh medium containing Asp8[H40-PEG@(RBC-H) nanoparticles and DiI-dyed RBC-H (with H40-PEG concentration at 100 μg mL-1). After 4 h, cells in all groups were washed twice using cold PBS and fixed using 4% paraformaldehyde (PFM) for 20 min at room temperature. Cold PBS was used to wash cell climbing slices for a total of three times. Nuclei for all cell lines were dyed with DAPI for 5 min and rinsed three times using cold PBS. Finally, all samples were detected with CLSM.
Cellular uptake of biomimetic NPs by macrophages
To evaluate macrophage uptake of different nanoparticles, CLSM and flow cytometry were used to determine immune escape abilities of resulting biomimetic NPs in vitro. Briefly, RAW 264.7 macrophage cells were seeded at a density of 1.0 × 105 cells well-1 into 6-well plates containing cell climbing slices and 1.5 mL of complete DMEM. Incubation was done at 37 ℃ for 12 h. Cells were incubated with FITC loaded H40-PEG NPs and FITC loaded Asp8[H40-PEG@(RBC-H)] NPs (FITC concentration at 10 μg mL-1) for 4 h at 37 ℃. Cells on the climbing slices were washed twice using cold PBS and fixed with 4% PFM for 20 min at room temperature. Then, the cells were rinsed three times using cold PBS. After cell nuclei had been stained with DAPI for 5 min, they were rinsed three times using PBS. Finally, slices were mounted and observed by CLSM. To quantify cellular uptake of the nanoparticles by RAW 264.7 cells, cells were digested with pancreatin after incubation for 4 h. Then, cells were obtained and centrifuged (3.0 ×103 rpm, 3 min, 4 ℃). Finally, harvested cells were suspended in cold PBS, and subjected to flow cytometry (CytoFLEX, Beckman Coulter) for analysis of fluorescence intensities of all groups.
Cellular uptake Asp8[H40-PEG@(RBC-H) NPs
The intracellular uptake assay was performed through CLSM and flow cytometry. Due to red fluorescence and hydrophobic small molecule characteristics, doxorubicin hydrochloride (DOX·HCl) was used as the model drug in the intracellular uptake experiment.
CLSM
To visualize the endocytosis process of biomimetic nanoparticles by WSU-HN6 cells, CLSM was used for imaging at preset time points. Briefly, WSU-HN6 cells were seeded in 6-well plates with cell climbing slices at 1.0 × 105 cells/well in 1.5 mL of complete DMEM and incubated for 12 h. Then, the complete medium was removed, and cells were incubated with Asp8[H40-DOX@(RBC-H)] nanoparticles (1 mL of DMEM medium) at a final DOX concentration of 10 μg mL-1. Cells were incubated for predetermined intervals at 37 ℃. Subsequently, they were washed twice using cold PBS and fixed in 4% PFM for 20 min at room temperature. Slices were rinsed three times using cold PBS. Afterwards, the nuclei of WSU-HN6 cells were stained with DAPI for 5 min. Slices were rinsed three times using PBS. Finally, slices were mounted and observed by CLSM.
Flow cytometry
WSU-HN6 cells were seeded into 6-well plates at 1.0 × 105 cells well-1 in 1.5 mL of complete DMEM and incubated for 12 h. Then, biomimetic NPs were dissolved in DMEM culture medium at a final DOX concentration of 10 μg mL-1. They were added into different wells and incubated for 5, 15, 30, 60, and 240 min, respectively. Thereafter, DMEM was removed, and cells were washed twice using cold PBS and treated with trypsin (200 μL). Subsequently, 2 mL of PBS was added to each well and the solutions were centrifuged at 4 ℃ for 5 min (3.0 ×103 rpm). After removal of the supernatants, 500 μL of cold PBS was used to resuspend the cells. Data for 1.0 × 104 gate events were collected and analyzed by flow cytometer (CytoFLEX, Beckman Coulter) and FLOWJO 10 software.
TEM and DLS assay
For optimal anticancer efficacy, photosensitizer IR780 and hypoxia-activated chemotherapy drug TPZ were selected model drugs in this study. To characterize Asp8[H40-TPZ/IR780@(RBC-H) nanoparticles, the size and ζ-potential of the nanoparticles were measured by Malvern Zetasizer Nano S in distilled deionized water at room temperature. Then, the size and morphology of the hybrid membrane-camouflaged nanoparticles were directly captured by TEM (Talos F200s instrument Thermoscientific, Czech).
TPZ/IR780 loading and release in vitro
The UV-vis absorption spectra were utilized to ensure successful encapsulation of IR780 and TPZ in Asp8[H40-PEG@(RBC-H) NPs. Standard curves for TPZ and IR780 were determined by UV-vis spectrophotometry. Drug loading and encapsulation efficiencies of both TPZ and IR780 were respectively calculated through the corresponding standard curve. Drug loading and encapsulation efficiencies were determined as:
Equation (2): Drug loading efficiency (DLE) = (Weight of the drug in resulting nanoparticles/Weight of the nanoparticles) × 100%
Equation (3): Drug encapsulation efficiency = (Weight of the drug in nanoparticles/Weight of the feeding drug) × 100%
Moreover, TPZ and IR780 release rates from Asp8[H40-TPZ/IR780@(RBC-H) NPs were determined using dialysis tubes containing PBS buffer (0.01M, pH=7.4) and acetate buffer (0.01M, pH=5.0), respectively. Briefly, Asp8[H40-PEG@(RBC-H) NPs (2.0 mg mL-1, 2.0 mL) were placed in dialysis tubes (MWCO=3500 Da) and soaked in 50 mL of both aforementioned buffer solutions, respectively. Then, they were placed in a 37 ℃ constant temperature shaker at 100 rpm. Dialysate (2.0 mL) was obtained at designated time points. Moreover, 2.0 mL of the fresh supplement buffer was added to replace the sampled buffer. TPZ or IR780 concentrations of samples were measured to determine release rates of corresponding drug contents through a UV-vis spectrophotometer. To investigate whether near infrared light-triggered the drug release, both buffer solutions were irradiated under 808 nm laser for 5 min (1.0 W cm-2) and 2.0 mL of solution was obtained at designated time points. The remaining process was performed as earlier described.
Hemolysis assay
To evaluate the safety of biomimetic NPs, a hemolysis experiment was performed. Briefly, fresh Balb/C blood was obtained in heparin sodium-containing tubes. RBCs were separated and collected by centrifugation at 3.0 × 103 rpm for 10 min at 4 ℃ and washed several times using ice-cold PBS. The process was repeated more than three times to make sure the supernatant was colorless. Then, the RBCs were diluted using ice-cold PBS to a concentration of 2% (v/v). Biomimetic nanoparticles and control polymers (Triton X-100 and Dextran) were prepared at serial concentrations (0.02, 0.05, 0.1, 0.2, 0.5, 1.0, and 2.0 mg mL-1) using PBS. Then, 0.5 mL of the biomimetic NPs and control polymers were added to 0.5 mL of the 2% (v/v) RBC solution with gentle shaking at 37 ℃. After 1 h of incubation, samples were centrifuged at 1.0 × 104 rpm for 10 min at 4 ℃, Supernatants (200 μL) were collected and transferred to 96-well plate. About 1% of triton X-100 solution (v/v, 100% lysis) was used as positive control while Dextran was utilized as the negative control. Hemoglobin absorbance was measured using a multi-functional microporous plate reader at a wavelength of 545 nm. Hemolysis degree was assessed as a mean of three independent experiments using the equation(4): The hemolysis percent = (ODsample-ODnegative)/(ODpositve-ODnegative) × 100%.
ROS/hypoxia detection in vitro
To assess the production of intracellular ROS/hypoxia, a ROS-ID® Hypoxia/Oxidative Stress Detection Kit (ENZO, USA) was used, according to the manufacturer’s instructions [46]. In briefly, WSU-HN6 cells were seeded into a laser confocal cell culture dish at a density of 5.0 × 104 per well in 1.0 mL of complete DMEM. After incubation for 12 h, cells were treated as follows: i. The medium was replaced with fresh completed DMEM (1.0 mL) containing PBS, which was used as negative control group; ii. Medium was replaced with fresh completed DMEM (1.0 mL) containing Asp8[H40-TPZ/IR780@(RBC-H)] NPs (50 μg mL-1 of IR780, according to IR780) for 4 h without or iii. with a laser irradiation (808 nm, 1.0 W cm-2, 5 min); iv. The medium was replaced with fresh completed DMEM (1.0 mL) containing an ROS inducer pyocyanin (Pyo), for 30 min or v. a hypoxia inducer, deferoxamine (DFO), for 3.5 h, respectively. Then, the cells were incubated with both hypoxia probes and ROS probes for 10 min and observed utilizing CLSM. The ROS green fluorescence and hypoxic red fluorescence signals were detected by CLSM with excitation wavelengths of 488 nm and 561 nm, respectively.
Cytotoxicity and antitumor efficacies in vitro
HUVEC cells and WSU-HN6 cells were respectively cultured in DMEM supplemented with 10% FBS, 100 U/mL streptomycin and 100 U/mL penicillin at 37 ℃ in a humidified incubator containing 5% CO2 (Thermo Scientific, USA). Relative cytotoxicity of Asp8[H40-PEG@(RBC-H)] biomimetic NPs against both cells were assessed through CCK-8 assay. Briefly, HUVEC cells and WSU-HN6 cells were seeded in 96-well plates at a density of 4.0 × 103 cells /well in 200 μL of complete medium for 24 h at 37 ℃. Then, the culture medium was removed and replaced with 200 μL of the medium containing serial of concentrations of the biomimetic nanoparticles. The final concentration was in the range of 5-40 μg mL-1. At least three wells were prepared for each dose of nanoparticles. These cells were incubated for another 48 h. The CCK-8 solution (10 mL) was added into each well, and the 96-well plates were incubated for 1 h at 37 ℃. Absorbance value for each well was measured using a multi-functional microporous plate reader (SpectraMAX iD5, USA) at a wavelength of 450 nm.
To assess the anticancer effects of biomimetic NPs in vitro, WSU-HN6 cells were seeded into 96-well plates at a density of 4.0 × 103 cells /well in 200 μL of complete medium. Subsequently, cells were allocated into two groups, including normoxia-therapy group and the hypoxia-therapy group. Cells in the hypoxia-therapy group were transferred from normoxic condition (20% O2) to hypoxic chamber (1% O2). Incubation was performed for an additional 12 h. Then, 200 μL of fresh medium containing different concentrations of biomimetic NPs were added to replace the spent medium (n=3 per group). After incubation in hypoxia or normoxia for 6 h away from light, cells were washed twice using cold PBS. Subsequently, the culture medium was replaced with PBS, and cells were either irradiated with the laser (808 nm, 1.0 W cm-2, 5 min) or not. After incubation for 24 h in normoxic conditions, the ability of WSU-HN6 cells was determined by performing the CCK-8 assay. Procedures for the CCK-8 assay as described above.
Calcein-AM/PI double staining assay
To assess the viability of WSU-HN6 cells in different nanoparticles, calcein-AM/PI double staining was performed and observed by fluorescence microscope (EVOS FL Auto, USA). Briefly, WSU-HN6 cells (5.0 × 104 cells well-1) were seeded in 12-well plates in 1.5 mL of complete medium. After culturing 12 h, different concentrations nanoparticles and PBS were added to the wells for 12 h. And cells in all groups were irradiated with 808 nm laser for 5 min, or not. Finally, calcein-AM/PI double staining was carried out, and fluorescence images of the live and dead cells were imaged by fluorescence microscope (EVOS FL Auto, USA).
Cell apoptosis in vitro
To determine the ability of various nanoparticles with and without laser, an Annexin V-FITC/PI apoptosis detection kit and flow cytometry were used in this study [47]. Briefly, WSU-HN6 cells were seeded into 6-well plates at a density of 5.0 × 104 per well in 1.0 mL of complete DMEM. After incubated for 24 h at 37 ℃, cells were grouped and treated differently, including with PBS, H40-PEG loaded IR780 NPs, Asp8[H40-IR780@(RBC-H)] NPs, and Asp8[H40-TPZ/IR780@(RBC-H)] NPs (10 μg mL-1 of IR780). The PBS group was set as the control. After incubation 4 h, suspensions of all groups were replaced with fresh complete DMEM. Cells in laser groups were irradiated with 808 nm laser for 5 min (1.0 W cm-2). Then, cells in all groups were cultured in fresh complete DMEM at 37℃ for another 4 h and treated with the apoptosis Analysis Kit. Finally, apoptotic cells were analyzed by flow cytometry (CytoFLEX, Beckman Coulter) and corresponding software.
Infrared imaging in vitro
To evaluate photothermal characteristics of biomimetic NPs in vitro, a thermal imaging camera (FOTRIC 365C, Shanghai, China) was used to obtain thermal images under irradiation using an 808 nm laser for 5 min with serial power densities (0.5 W cm-2, 0.6 W cm-2, 1.0 W cm-2, 1.2 W cm-2, 1.5 W cm-2). Biomimetic NPs with different IR780 concentrations (0, 50 μg mL-1, 100 μg mL-1) in 12-well plate were further irradiated with the 808 nm laser (1.0 W cm-2, 5 min).
Infrared imaging in vivo
To evaluate photothermal effects of the biomimetic NPs, an infrared thermal camera was used to determine effective therapy in vivo [48]. Briefly, tumor-bearing mice were intravenously injected with Asp8[H40-TPZ/IR780@(RBC-H)] NPs (1.6 mg Kg-1 of IR780 dose) or normal saline. Then, 8 h after injection, mice were irradiated by laser for 5 min (808nm, 1.0 W cm-2). The Fotric AnalyzIR software was utilized to analyze the thermographs.
Biodistribution in vivo
To assess nanoparticle biodistribution in bone invasion models, DiD, a lipophilic near-infrared fluorescent dye, was used to mark different nanoparticles [49]. Tumor-bearing nude mice were intravenously administered with serial DiD-loaded nanoparticles or normal saline through the tail vein at a prescribed DiD dose (50 μg mL-1). Then, 8 h after injection, mice were sacrificed, and the main organs, including the heart, liver, spleen, lung, kidney, the lesion and healthy right mandibula were carefully dissected, rinsed using normal saline, and stored away from light for further analysis. DiD fluorescence visualization of each organ (Ex: 655 nm, Em: 714 nm) was performed using an imaging system (VISQUE In Vivo Smart).
Therapeutic evaluation of Asp8[H40-TPZ/IR780@(RBC-H) in vivo
To evaluate anticancer efficacy of various TPZ/IR780-loaded NPs in a bone invasion model, mouse model whose right mandibular had been invaded were selected as experimental animals. Model were developed as previously reported with minor alterations [50]. Briefly, 2.0 × 106 WSU-HN6 cells were injected into the right masseter region of female BALB/c nude mice (4-6 weeks old). After 1 week, tumor formation rates were determined to be 100%. Then, mice were randomly assigned into 7 groups (five mice per group), including, i. Normal saline, ii. Normal saline+laser, iii. TPZ+laser, iv. Asp8[H40-PEG@(RBC-H)] NPs+laser, v. H40-PEG loading IR780 NPs+laser, vi. [H40-IR780/TPZ@(RBC-H)] NPs+laser, and vii. Asp8[H40-IR780/TPZ@(RBC-H)] NPs+laser. Mice in all groups were administrated with 100 μL PBS or different formulations at a TPZ dose of 1.5 mg Kg-1 and a IR780 dose of 1.6 mg Kg-1 through the tail vein. Mice in the PBS group were used as negative controls. The first day of administration was designated as day 0. Tumor volumes and body weights of mice were measured every day. After 24 h, mice in the laser group were illuminated with 808 nm wavelength laser irradiation (1.0 W cm-2, 5 min). Moreover, treatment and laser irradiation were repeated on day 2, 4, 6, 8, 10, 12, 14, 16. Tumor volumes (V) were determined using the equation: V= Width2 × Length/2. According to ethical reasons, mice with tumor sizes exceeding 150 mm in any dimension were sacrificed. Otherwise, mice were euthanized on day 16, tumors were dissected, weighted, photographed, and fixed in 4% paraformaldehyde for further analysis. Hematoxylin and eosin (H&E) staining and fluorescein (FITC) terminal deoxynucleotidyltransferase-mediated UTP end labeling assay (TUNEL, Servicebio, China) were used to evaluate the changes of cancer tissue and to detect apoptosis in tumor slices.
Micro-computed tomography reconstruction of right mandibula
To assess the degrees of mandibula destruction by cancer, micro-computed tomography (micro-CT) scanning was used to scan the right mandibula of all mice. Mice were euthanized on day 16 after which tumor-bearing mandibula were carefully dissected and placed into 4% PFM solution. Afterwards, each specimen was scanned using a micro-CT scanner (Scanco VivaCT40, Switzerland). Three-dimensional models were reconstructed using a Scanco VivaCT40 micro-CT software.
Systemic toxicity evaluation
To assess the biosafety of biomimetic NPs during therapy, besides measuring body weights of mice every day, changes in blood and major organs are also important indicators [51]. Mice were sacrificed after 16 days of treatment. Blood samples were obtained from the orbital venous before sacrifice and analyzed at the stomatological hospital of Chongqing medical university. Moreover, major organs including the heart, liver, spleen, lung, and kidney were carefully dissected and fixed with 4% PFM for further H&E staining and analysis.
Statistical methods
Unless otherwise stated, statistics were determined via a one-way ANOVA with Tukey multiple comparisons test using GraphPad Prism Version 8.0.1 software. All reported data show means and standard error unless otherwise noted. * P < 0.05, ** P < 0.01, and *** P < 0.001 were determined statistically significant with n ≥ 3 for all experiments.
{kind=link}
{kind=link}