We identified tandem repeats in a human reference genome (GRCh38) using tantan [23] (http://cbrc3.cbrc.jp/~martin/tantan/). In total, 3,347,418 loci were identified, with the repeat units ranging from 1 to 2000 bp. We used 21 publicly available long read whole genome sequencing datasets (we suppose they do not have pathogenic tandem repeat expansions), with average coverage of 27x (ranging 8x-48x, Table S1). tandem-genotypes predicted lengths for more than 98% of the 3 million tandem repeats (Table S1), including 215,561 triplet repeats.
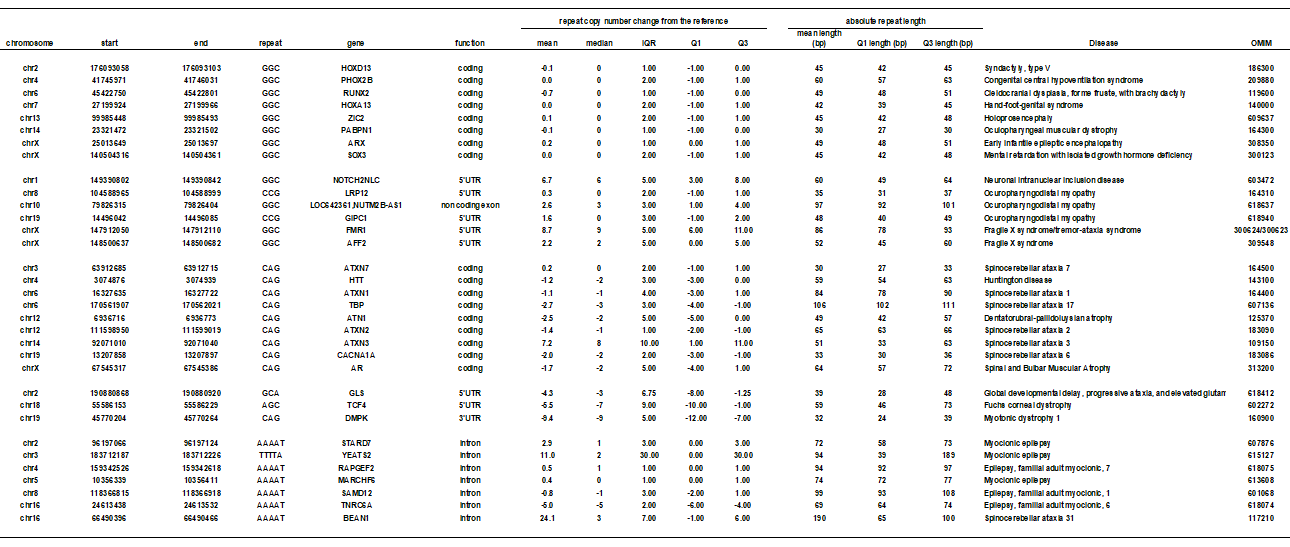
We investigated 12 CAG and 14 GGC triplet repeat and 7 AAATA quintuplet repeat disease loci (Table 1), and plotted the distribution of copy number changes from the reference in all the reads. We found that disease-causing repeats show different distribution from other non-disease repeats (Supplementary Fig S1A-C). We randomly extracted the same number of non-disease repeat loci for comparison to the disease repeat loci (CAG: n = 12, GGC: n = 14, AAAAT: n = 7)) (Supplementary Figure S1). This supports our hypothesis that disease-causing tandem repeats are more polymorphic among the normal population than other loci.
Given that different repeat sequences may have different mutation rates [24], we compared the ten kinds of non-disease triplet repeats (All triplet repeats can be categorized into 10 kinds. Note that AAC repeats includes AAC, ACA, CAA, GTT, TGT, TTG repeats) (Supplementary Figure S2). We plotted the variation of repeat length (interquartile range (IQR) of repeat-unit count from each read), and mean repeat length, at each exonic locus (including UTR). Most of the non-disease triplet repeats have little or no length polymorphism. A large fraction (> 94% of all repeats) have IQR 2 or less, while disease causing tandem repeats usually show more variation (always more than 2) (Table 1). It is of interest that GGC and CAG repeats have more polymorphic loci than other repeat structures (Supplementary Figure S2). In addition, shorter-unit repeats are more numerous and more variable (Supplementary Figure S3). Therefore, we analyzed the variation (IQR) and repeat length for disease causing repeats in comparison to other repeats considering the repeat unit and repeat location.
Disease-associating CAG repeats are longer and more variable than most other CAG repeats (Fig. 1A, B, Table 1). We showed coding and non-coding repeats separately (A: coding, B: non-coding). All disease-causing CAG repeats are located in protein-coding regions except for DMPK, GLS, and TCF4 which are in 5’UTR (Table 1). Next we tested GGC repeats. Disease-causing 5'-UTR GGC loci are long and variable (Fig. 2B) but protein-coding regions are long but show less variability (Fig. 2A). Gene names were used to indicate the disease-causing repeats because the pathogenic repeats are present only once in each gene. All known protein-coding GGC repeat diseases are located at poly-alanine tracts. This may reflect the difference in disease mechanisms of protein-coding versus 5'-UTR GGC repeats or protein-coding GGC versus CAG repeats. Next, we examined the variation and length of all intronic AAAAT repeat loci in 21 individuals, and found several highly polymorphic AAAAT repeats including disease loci (Fig. 3, Table 1).
We repeated our analysis using repeat annotations from Tandem Repeats Finder (TRF, a.k.a. simpleRepeat.txt) [25]. TRF annotates fewer repeats than tantan (Supplementary Figure S4A), however, the proportion of triplet repeat sequences is similar (Supplementary Figure S4B). Numbers of intersections between these annotations were calculated using bedtools v2.27.1 (Supplementary Table S2). We analyzed disease-associated CAG and GGC repeats, and observed similar results to tantan-annotated repeats (Supplementary Figure S5: CAG, S6: GGC, S7: AAAAT).
Next, we tested if polymorphic disease-associated tandem repeats are correlated with reported GWAS SNPs. We tested ATXN3 and GLS disease-associated repeats because they are highly polymorphic among disease-associated CAG repeats. These repeats have two (rs12588287: coronary artery calcification [26], rs10143310: ALS [27]) and one (rs4853525: reticulocyte count [28]) near-by GWAS SNPs (< 10 kb) [22], respectively. Due to the limited coverage and read length, we could obtain genotypes in most but not all of the 21 cases (Supplementary Table S3). In each case, one of the two SNP alleles is significantly (p < 0.05, unpaired t-test) associated with longer repeats (Supplementary Figure S8). Risk alleles tend to occur with shorter repeats for two SNPs: rs4853525-C and rs12588287-T. Risk allele for rs10143310 is not available [27]. This merits further investigation by genotyping a larger number of individuals.
Finally, we listed highly polymorphic repeats (IQR > = 5) which have very near GWAS signals (< 100 bp) from a GWAS catalog [22] (Table S4). We found an interesting candidate, an intronic repeat in the CLN8 gene: a SNP within this repeat (rs11986414) and a near-by SNP (rs4875960) are reported to be associated with severity of Gaucher syndrome [29]. It is an intriguing possibility that this repeat genuinely acts as a driver of the GWAS signals and affects the disease severity. We found that the A genotypes of these two SNPs are correlated with shorter repeat (Supplementary Figure S9). It would be interesting to investigate functional consequences of changing these repeats. These speculative examples need further association studies targeting near-by tandem repeats together with functional studies to elucidate the mechanistic relation to the phenotype.
{kind=link}