Genome-wide examination of cytosine methylation in ‘Qinguan’ and ‘Nagafu No. 2’ axillary bud
About 118.36 and 120.35 million raw reads of two BS libraries for FA and QA were generated by the Illumina HiSeq 2000 platform, respectively (yielding ~ 35.51 and ~ 36.11 data for FA and QA) (Table S1). Additionally, 114,420,604 and 118,150,703 clean reads for FA and QA were produced, respectively, as well as 62,656,722 (54.76%) for FA and 73,170,730 (61.93%) for QA clean reads were uniquely mapped to the Malus x domestica reference genome (https://www.rosaceae.org/species/malus/malus_x_domestica/genome_GDDH13_v1.1) [36] (Table S2). The detail information of gene coverage for FA (76.4%, 63.12% and 20.42%) and QA (77.11%, 63.62% and 18.78%) involved in CG, CHG and CHH contexts, respectively, were indicated in Table S3.
The circus plots showed that the whole chromosome-wide distribution of DNA methylation involved in the density of 5mC in the CG, CHG, and CHH contexts, the density of TEs, the genes and RNAs density of each chromosome for QA and FA (Fig. 2A, B; Fig. S2), indicating that there were significant differences for these indicators among various chromosomal positions. Additionally, there were similar proportion of methylation levels in CG, CHG and CHH contexts for FA (i.e., 19.35%, 62.96% and 17.68% for CG, CHG and CHH, respectively) and QA (19.64%, 62.49% and 17.86% for CG, CHG and CHH, respectively) (Fig. 2C). The detail information of methylated CX percentage for FA and QA involved in CG, CHH and CHG contexts in each chromosome were showed in Fig. 2D, indicating that the percentage of CG context was significantly higher than CHG and CHH for both FA and QA. However, the mean methylation levels involved in CG, CHG and CHH contexts were significantly higher in QA than in FA (Fig. 2E). Bisulfite sequencing data for the mean depth and proportion of covered bases in each chromosome for the QA and FA were in Fig. S3. The methylation densities, and levels and in the all contexts in QA and FA were in Fig. S4.
Distribution of DNA methylation in genic regions
A heat map analysis showed that DNA methylation levels were significantly differences among different gene features including promoter, exon, intron, and repeat regions (Fig. S5A). The DNA methylation levels including CG, CHG and CHH contexts for both FA and QA were highest in the repeat regions, followed by the promoter, exon, and intron regions (Fig. S5A, B). Additionally, the DNA methylation levels in all contexts were higher in the regions 2-kb upstream and downstream of the gene-bodies than in the gene-bodies (Fig. S5C).
Circos plots showed that there were significant differences in CG, CHG and CHH contexts in the whole chromosome-wide distribution of DNA methylation levels between FA and QA (Fig. 3A). DNA methylation levels including the CG, CHG, and CHH contexts were similar between FA and QA in all the different gene regions (i.e., promoter, exon, intron, and other regions) (Fig. 3B), as well as the genebody and the regions 2-kb upstream and downstream of the gene-bodies (Fig. 3B). Specifically, the DNA methylation levels in exon region for CG and CHG contexts were significantly higher in QA than in FA (Fig. 3B), but the DNA methylation levels in promoter region for CHH, and in intron region for CHG and CHH contexts, as well as in repeat region for CHH context had an opposite results (Fig. 3B). Additionally, for CHH context in the genebody and the regions 2-kb upstream and downstream of the gene-bodies regions, the DNA methylation levels in QA were significantly lower than in FA (Fig. 3C), but for CG context in genebody region had an opposite result between QA and FA (Fig. 3C).
Identification of differentially methylated regions (DMRs) between axillary bud with profusely flowering ‘Qinguan’ and no flowering ‘Nagafu No. 2’, and functional annotation analysis
Circos plots showed that the whole chromosome-wide distribution of differentially methylated regions (DMRs) (FDR < 0.05) for both hypermethylated and hypomethylated in CG, CHG and CHH contexts between FA and QA (Fig. 4A; Fig. S6). Venn diagrams present unique or public DMRs in genes (QA_vs_FA_DMRs_genes) and in promoter regions (QA_vs_FA_DMRs_promoter_genes) between FA and QA in the CG, CHG, and CHH contexts (Fig. 4B). Specifically, the QA_vs_FA comparison group comprised 2820, 1426, and 1808 DMR_genes (Fig. 4B) as well as 2853, 2462, and 2547 DMR_promoter_genes in the CG, CHH, and CHG contexts, respectively (Fig. 4B). Additionally, the DMR methylation levels in the CG and CHG contexts of the DMRs were significantly higher in QA than in FA, but the DMRs in CHH contexts had an opposite result between FA and QA (Fig. 4C). A heat map also showed the visual information of the DMRs and their DNA methylation levels in all three contexts (FDR < 0.05) between FA and QA (Fig. 4D). The numbers of the hypermethylated or hypomethylated DMRs in all contexts also had a big difference among different genic regions (i.e., the promoter, TSS, TTS, exon, repeat, and other regions) (Fig. 4E). For example, the numbers of DMRs (i.e., hypermethylated or hypomethylated) in TSS, TTS, utr5, and utr3 regions were significantly less than other regions including promoter, exon, repeat (Fig. 4E). Additionally, the numbers of hypermethylated DMRs in CG and CHG contexts in repeat region were significantly more than hypomethylated DMRs between FA and QA, but for CHH context, there was an opposite result (Fig. 4E).
To know the function of the DMR_genes and DMR_promoter_genes, the most enriched GO terms were identified for both hypermethylated and hypomethylated in QA compared with FA involved in CG, CHG and CHH contexts (Fig. S7, S8). For example, the most enriched GO terms included metabolic process, cellular process, biological regulation for QA_vs_FA_hypermethylated in the CG context (Fig. S7). For the DMR_promoter_genes, metabolic process, cellular process, biological regulation in biological process, cell, cell part, and organelle in cellular component, binding, catalytic activity, transporter activity in molecular function were the most enriched GO terms for QA_vs_FA_hypermethylated in the CG context (Fig. S8). The KEGG pathway also revealed some significantly enriched pathways involved in the DMR_genes and DMR_promoter_genes for the QA_vs_FA comparison group (Fig. S9, S10), including the following: metabolic pathways, biosynthesis of secondary metabolites for hyper/hypo_DMR_genes in the CG context (Fig. S9A, D). Additionally, the information regarding the significantly enriched pathways for the Hyper/Hypo_DMR_promoter_genes in the CG, CHG, and CHH contexts was provided in Fig. S10A–F.
Transcriptome profiling and functional analysis of differentially expressed genes between axillary bud with profusely flowering ‘Qinguan’ and no flowering ‘Nagafu No. 2’ apple
The hierarchical clustering heat maps showed the differentially expressed genes in the QA_vs_FA comparison group including 19,656 (9,543 downregulated and 10,113 upregulated) DEGs (log2 fold-change > 0; padj < 0.05) (Fig. 5A, B). The GO functional analysis revealed the most enriched terms for the DEGs involved in biological process (i.e., oxoacid metabolic process, organic acid metabolic process, and catabolic process for upregulated genes; as well as signaling, signaling transduction, and single organism for downregulated genes), cellular component (i.e., membrane protein complex, thylakoid, thylakoid part for upregulated genes; as well as cell periphery, cell cortex, plasma membrane part for downregulated genes) and molecular function (i.e., peptidase activity, endopeptidase activity, transferase activity for upregulated genes; as well as calcium ion binding, ubiquitin-protein transferase activity, calmodulin binding for downregulated genes) in QA than in FA (Fig. 5C). Additionally, plant-pathogen interaction, spliceosome, amino sugar and nucleotide sugar metabolism for the upregulated genes, as well as carbon metabolism, biosynthesis of amino acids, protein processing in endoplasmic reticulum for the downregulated genes in QA than in FA were the most enriched KEGG pathways (Fig. 5D). Additionally, the gene expression distribution, as well as pearson correlation among samples of QA and FA were in Fig.S11. Details regarding the RNA-sequencing data for both QA and FA were in Table S4, S5.
Association between DNA methylation and transcript abundance
The gene expression levels were divided into the following four quartiles based on their FPKM values: non-expressed (FPKM < 1), low expression level (1 ≤ FPKM < FPKM_25%), medium expression level (FPKM_25% ≤ FPKM < FPKM_75%), and high expression level (FPKM ≥ FPKM_75%) (Fig. 6A). We found that the lowly expression genes had high methylation levels, as well as highly expression genes had low methylation levels in the gene-body for CG, CHG and CHH contexts, and upstream of the TSS and downstream of the TTS regions for CG and CHG contexts for both QA and FA (Fig. 6A). However, for CHH context, the most highly expression genes had the high methylation levels in upstream of the TSS and downstream of the TTS regions (promoter regions) in both QA and FA (Fig. 6A).
To know the correlation between the gene methylation and expression levels, the methylated genes were divided into the following five quintiles based on the promoter and gene-body methylation levels: group 1 (methyl_level < level_20%), group 2 (level_20% ≤ methyl_level < level_40%), group 3 (level_40% ≤ methyl_level < level_60%), group 4 (level_60% ≤ methyl_level < level_80%), and group 5 (methyl_level ≥ level_80%) (Fig. 6B). We also found that methylation levels had a negative regulatory relationship with gene expression levels involved in the CG, CHG, and CHH contexts in the gene-body region, and CG and CHG contexts in upstream of the TSS and downstream of the TTS regions (promoter regions), but they had a positive regulatory relationship when the methylation in CHH context happened in promoter regions (upstream of the TSS and downstream of the TTS regions) for both QA and FA (Fig. 6B). Additionally, the detail information about the relationship between methylation difference and gene expression (FPKM) involved in CG, CHG and CHH contexts for both hypermethylated and hypomethylated DMRs in different regions (i.e. gene-body and promoter) in QA compared with FA were presented in Fig. 7A, B, C.
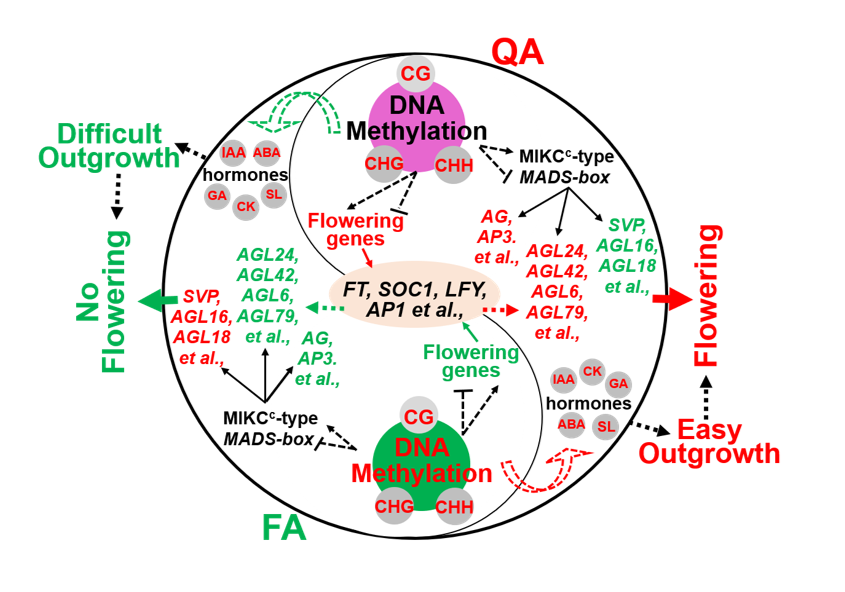
Huge differences in transcription of MIKCc-Type MADS-box genes by changing DNA methylation, contributed to contrasted flowering behaviors
We identified 132 MADS-box family genes in apple that belong to five subgroups (i.e., MIKCc, Mβ, Mα and Mδ) (Fig. 8A; Fig. S12). Among them, these differentially expressed MADS-box family genes were mainly belonged to MIKCc subgroup between QA and FA, but no differentially expressed MADS-box genes were found in the other two subgroups (Mα and Mδ) (Fig. 8A). Eleven differently expressed MADS-box genes belong to cluster I (i.e., AGL16_MD08G1206400; AGL18_MD00G1106300; SEP1_MD06G1204100/MD09G1009100; SEP4_MD13G1059300) were significantly lower expression in axillary bud with profusely flowering ‘Qinguan’ than ‘Nagafu No. 2’, but other 17 differently expressed MADS-box genes belong to cluster II (i.e., six AGL24, three AGL80, AGL6, AGL79 and AGL24) had an opposite result (Fig. 8B; Additional file 1). Specially, flowering key genes (SOC1_MD07G1123600 and AP1_MD06G1204400), as well as floral meristem identify genes (AG_MD10G1271000 and AP3_MD02G1136500) were belong to cluster II that showed higher expression levels in axillary bud with profusely flowering ‘Qinguan’. However, the key negative regulation gene in floral induction (SVP_MD01G1038600/MD15G1384600) belong to cluster I were significantly lower expression in axillary bud with profusely flowering ‘Qinguan’ than ‘Nagafu No. 2’ (Fig. 8B; Additional file 1). Additionally, the methylation levels of thirteen MADS-box family genes (i.e., AGL21_intron_CHG, AGL21_promoter_CHH and AGL12_intron_CHH and AGL62_exon_CHG) in different gene regions were significantly higher expression in axillary bud with profusely flowering ‘Qinguan’ than ‘Nagafu No. 2’ (Fig. 8C), which were contributed to down-regulation of these genes in axillary bud of ‘Qinguan’ than in ‘Fuji’. However, other eleven DMRs (i.e., AGL30_exon_CG, AGL42_promoter_CG, and AGL42_promoter_CHG) had an opposite methylation pattern between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ apple, which may contribute to the regulation of these MADS-box genes expression involved in promoting flowering in ‘Qinguan’ axillary bud (Fig. 8C). Similarly, IGV snapshots presented the representative DMRs that were hypermethylation or demethylation between the axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’, including AGL80_MD00G1098000_CHH, AGL42_MD01G1103300_CG, and AGL42_MD00g1004500_CG in the promoter, and AGL79_MD17G1001300_CHH in the intron, and AGL30_MD13G1070300_CG in the exon (Fig. 8D). Additionally, multiple differently expression of MADS-box genes formed complex gene interaction regulatory networks including floral promoter (i.e., SOC1 and AGL42), floral organ identify (i.e., AG and AP3), and floral repressor (i.e., SVP and AGL18), contribute to regulation of flowering in axillary buds of ‘Qinguan’ (Fig. 9).
Differentially expression genes in multiple flowering pathways between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ apple by changing DNA methylation, contributed to contrasted flowering behaviors
We identified these different expression genes (DEGs) and different methylation regions (DMRs) in multiple flowering regulating pathway between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ apple (Fig. 10; Additional file 2, 3). Firstly, these genes involved in key floral meristem identify (i.e., LFY, AP1, SOC1 and FT) were significantly higher expression in axillary buds of ‘Qinguan’ than in ‘Nagafu No. 2’ (Fig. 10A). In age pathway, eleven SPL genes that can directly up-regulated LFY and SOC1 showed significantly higher expression in ‘Qinguan’ axillary buds than in ‘Nagafu No. 2’, but there had an opposite result in two AP2-like and one TOE genes involved in negative regulation of SOC1 expression (Fig. 10A). In photoperiodic pathway that centered on the CO gene, seven COL and eleven BBX genes were significantly lower expression in ‘Qinguan’ axillary buds than in ‘Nagafu No. 2’, and the upstream regulator of the CO gene, such as five COPs, three SPAs, PHYA, CRY1 and CRY2 had a similar result (Fig. 10A). And another upstream regulator of CO gene involved in circadian clock pathway including FKF1, two CDF2 and two CDF3, three PRR and TOC1 also had a similar result (Fig. 10A). However, two NF-YA, four NF-YB and four NF-YA that can interact with CO gene were significantly higher expression in axillary buds of ‘Qinguan’ than in ‘Nagafu No. 2’ (Fig. 10A). In vernalization and autonomous pathway, these genes were significantly upregulated in ‘Nagafu No. 2’ compared with ‘Qinguan’ including two MSI1, SWN and VIN3, as well as two FLK and FY that can directly inhibit the expression of FLC gene to promoting flowering (Fig. 10A). Additionally, FAF1-like complex including ELF5, EFS, ATX2, three SDG2, two VIP4 and VIP5, and SWR1/SRCAP-like complex including ARP5, ARP6, SEF and PIE1 that can directly promote the expression of FLC gene to inhibit flowering were significantly higher expression in ‘Nagafu No. 2’ than in ‘Qinguan’, that contributed to promote floral induction in ‘Qinguan’ (Fig. 10A). And these genes including REF and SRP1 involved in histone modification were significantly differentially expression between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ (Fig. 10A), suggesting they play key role in regulation of flowering process in ‘Qinguan’. In sugar pathway, two types of genes (i.e., IDD4/5 and SUS4) belong to the upstream of FT and SPL genes were significantly higher expression in axillary buds of ‘Qinguan’ than in ‘Nagafu No. 2’ (Fig. 10A). And five TPS14 genes showed higher expression in QA than in FA, but one TPS2 and TPS4, as well as five TPS21 genes had an opposite result (Fig. 10A), indicating that TPS genes play multifaceted functions on flowering in sugar pathway. In addition, two RGA2 genes in GA pathway were significant differential expression between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’, contribute to both different flowering ability (Fig. 10A).
We also identified many different methylation regions (DMRs) between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ involved in multiple flowering pathways (Fig. 10B). For example, five DMRs(i.e., LFY_MD14G1146700_CG in promoter, and ELF4_MD11G1074300_CHH in exon)belong to cluster I were significantly lower expression in axillary bud with profusely flowering ‘Qinguan’ than ‘Nagafu No. 2’ in flowering pathways (Fig. 10B), but sixteen DMRs (i.e., SPL1_MD16G1137700_CHH, and SLP14_MD14G1060299_CHH in promoter; TFL1_MD12G1023900_CG in promoter and TFL2_MD07g1081800_CG in exon) belong to cluster II were had an opposite result (Fig. 10B). In photoperiodic pathway, twenty-one DMRs belong to cluster II, such as COL4_MD16G1148500_CG, and CRY2_MD16G1217800_CHG, DOF_MD15G126570_CG/CHG in exon, BBX32_MD17G1147000_CG/CHG in exon showed significantly higher methylation levels in QA than in FA (Fig. 10B), but BBX32_MD17G1147000_CHH in promoter, and COP9_MD09G1015400_CG in exon belong to cluster I had an opposite result, suggesting that these DMRs in different regions may contribute to regulate genes expression levels. Similarly, six DMRs (i.e., SPA3_MD11G1247500_CG in exon and SDG2_MD17G1091000_CHG in promoter) belong to cluster I were showed significantly lower levels in QA than in FA, and nineteen DMRs (i.e., ARP4_MD03G1252000_CG in exon, and VIN3_MD08G1199400_CG in promoter ) belong to cluster II had an opposite result (Fig. 2B). In sugar and GA pathways, five TPS21_MD03G1214900_CHG in intron, GRA_MD13G1022100_CG/CHG in exon, and _CHH in promoter, and TPS21_MD03G1207000_CHH in exon belong to cluster I were significantly higher levels in FA than in QA, but these DMRs belong to cluster II (i.e., IDD5_MD16G1009600_CHH in promoter, and GAI_MDMD17G1260700_CHG in exon, and _CG in promoter) had an opposite result (Fig. 10B).
Additionally, IGV snapshots showed these were multiple DMRs in gene different regions between QA and FA that affecting gene expression, such as hypermethylation in QA than in FA (i.e., SPL14_CHH, CRY1_CHH in promoter, PHYB_CG, and TPS21_CHG in exon, as well as BBX32_MD17G1147000 involved in CG, CHH and CHG contexts in exon region) or demethylation in QA than in FA, such as NF-YB_MD00G1101600 involved in CG, CHH and CHG contexts in exon region (Fig. 10C), suggesting that gene expression were regulated by these DMRs in different regions. We also found that these DMRs in different regions involved in CG, CHH and CHG contests were negatively correlated with gene expression levels, such as BBX32_CG _exon, COL6_CG_exon, NF-YB_CG_exon, and COL14_CG_promoter (Fig. 10D), but DMRs in promoter involved in CHH context can promote gene expression, such as SPL14_MD14G1020600_CHH in promoter (Fig. 10D), indicating that DNA methylation can positively or negatively regulate gene expression.
Significant differences in multiple hormone levels were due to DEGs and their DMRs in their synthesis pathways, leading to both contrasted axillary bud outgrowth and flowering behaviors.
There were significant differences in multiple plant hormones between axillary buds of ‘Qinguan’ and ‘Nagafu No. 2’ (Fig. 11A; Additional file 4). The ABA and GA contents were significantly higher in QA than in FA, as well as no significant difference in IAA, but content of CK had an opposite result between them (Fig. 11A), associated with the significantly higher rates of germination and flowering in QA than in FA (Fig. 11A).
In addition, these genes involved in ABA biosynthesis, such as ZEP, three NCED1, two NCED4, two ABA2 were significantly higher in axillary buds of ‘Nagafu No. 2’ than in ‘Qinguan’, a similar result about these key GA biosynthesis genes (i.e., two CPS1 and KS) can be found in our data (Fig. 11A), but an opposite result involved in these genes (i.e., GA2OX1, GA2OX2, and GA20X8) that changing active GAs into inactive GAs (Fig. 11A), contributing to lower GA levels in QA than in FA. We also found that IPT2 gene involved in CK biosynthesis, and YUCC8 involved in IAA biosynthesis were significantly higher expression in QA that in FA, that may lead to higher level of CK in QA than in FA (Fig. 11A). Interesting, these genes involved in SL biosynthesis (i.e., D27, MAX3, MAX4, and MAX1) were no significantly difference between QA and FA (Fig. 11A), but the genes belong to SL signaling including three D14 and two MAX2 were significantly higher expression in FA than in QA (Fig. 11A) involved in regulation of axillary bud outgrowth and formation through targeted regulation of BRC1 as well as it is regulated by auxin. Additionally, multiple PIN genes (i.e., two PIN1, PIN3, and five PIN-like) involved in IAA transport from axillary bud into other organs were significantly higher in QA than in FA (Fig. 11A), that making relative lower IAA level in axillary bud of ‘Qinguan’ to induce axillary bud outgrowth compared with ‘Fuji’.
Additionally, IGV snapshots showed GA2OX8_MD05G1341000 in CG, CHG and CHH in exon region involved in GA biosynthesis were demethylation in QA than in FA (Fig. 11B) associated with relative higher expression of GA2OX8 in QA, indicating that negative correlation between DNA methylation and gene transcription level. Additionally, the PIN1_MD12G1095100 in CHH in promoter region were also significantly higher in FA than in QA, but expression level of PIN1 had an opposite result between FA and QA (Fig. 11B), suggesting that DNA methylation play key roles in gene expression.
{kind=link}