Patient characteristics. Eighty-one patients with stage III and IV cancer were consecutively recruited based on the inclusion criteria. The clinical characteristics of the patients were shown in Supplementary Table S1. Based on the histology, 80% and 20% of the patients presented with Type I (mainly endometrioid) and Type II (non-endometrioid) tumors, respectively. Furthermore, the patients were grouped based on the clinical outcome as follows (Table 1): no evidence of disease and progressive disease (PD). The clinical characteristics of the patients in the two groups are shown in Table 1. No significant differences in histology, grading, and treatment were observed between the two groups of patients.
Table 1
| Characteristics | No evidence of disease (N = 47) | Progressive disease or death (N = 34) | P |
| Age (years) | | | 0.5287 |
| Mean (range) | 53.2 (34 ~ 76) | 58.8 (34 ~ 87) | |
| FIGO staging | | | <0.001 |
| Stage III | 44 (93.6%) | 18 (52.9%) | |
| Stage IV | 3 (6.4%) | 16 (47.1%) | |
| Histology | | | 0.151 |
| Endometrioid | 41 (87.2%) | 24 (70.6%) | |
| Others | 6 (12.8%) | 10 (29.4%) | |
| Grading* | | | 0.078 |
| 1 | 10 | 3 | |
| 2 | 23 | 13 | |
| 3 | 14 | 18 | |
| Survival (months) | | | |
| Progression-free survival | 48.4 (1.7 ~ 119.1) | 14.8 (1.7 ~ 75.5) | <0.0001 |
| Overall survival | 48.4 (1.7 ~ 119.1) | 23.9 (1.7 ~ 92.5) | <0.002 |
| Treatment | | | |
| Staging surgery | 47 | 31 | 0.834 |
| Others# | 0 | 3 | |
| Chemotherapy | 46 | 34 | 0.853 |
| *Pathological grading: clear cell carcinoma, serous carcinoma, and carcinosarcoma were classified as grade 3 |
| #Others: dilatation & curettage for diagnosis alone |
Clinical impacts of gene mutation. The Oncomine Comprehensive Assay v1, a targeted NGS assay, was applied to the FFPE tumor samples to detect the presence of mutations across 143 genes. Associations between the individual mutated gene and the clinical outcome with FDR adjusted p-value < 0.1 are shown in Tables 2 and 3. The mutation status of eight genes (MET, U2AF1, BCL9, PPP2R1A, IDH2, CBL, BTK, and CHEK2) were positively correlated with poor progression-free survival (PFS) and overall survival (OS). In contrast, IFITM1 and DNMT3A mutations were associated with better clinical outcomes. MET was selected for further studies because the adjusted p-value was the smallest and the frequency of mutation was not low.
Table 2
The association between the mutated genes and overall survival.
| Gene | Case number with mutation | Case number without mutation | p value | Adjusted p value | Hazard ratio | 95% interval (Lower ~ Upper) |
| MET | 24 | 57 | 0.019 | 0.067 | 2.606 | 1.167–5.819 |
| U2AF1 | 3 | 78 | 0.006 | 0.067 | 5.942 | 1.683 ~ 20.980 |
| BCL9 | 4 | 77 | 0.022 | 0.067 | 4.343 | 1.241 ~ 15.201 |
| PPP2R1A | 8 | 73 | 0.020 | 0.067 | 3.272 | 1.204 ~ 8.892 |
| IFITM1 | 79 | 2 | 0.018 | 0.067 | 0.151 | 0.031 ~ 0.724 |
| IDH2 | 2 | 79 | 0.018 | 0.067 | 6.44 | 1.381 ~ 30.036 |
| EGFR | 7 | 74 | 0.041 | 0.088 | 2.785 | 1.040 ~ 7.457 |
| CBL | 2 | 79 | 0.039 | 0.088 | 4.856 | 1.086 ~ 21.703 |
| DNMT3A | 9 | 72 | 0.049 | 0.088 | 0.222 | 0.049 ~ 0.995 |
| BTK | 2 | 79 | 0.039 | 0.088 | 4.856 | 1.086 ~ 21.703 |
| RB1 | 21 | 60 | 0.079 | 0.094 | 2.012 | 0.923 ~ 4.382 |
| CHEK2 | 4 | 77 | 0.067 | 0.094 | 3.215 | 0.921 ~ 11.230 |
| MLH1 | 14 | 67 | 0.077 | 0.094 | 2.216 | 0.918 ~ 5.348 |
| PIK3R1 | 37 | 44 | 0.065 | 0.094 | 2.113 | 0.954 ~ 4.680 |
| ESR1 | 2 | 79 | 0.058 | 0.094 | 7.715 | 0.937 ~ 63.546 |
| KRAS | 37 | 44 | 0.068 | 0.094 | 2.059 | 0.949 ~ 4.469 |
| FGFR3 | 7 | 74 | 0.083 | 0.094 | 2.621 | 0.882 ~ 7.792 |
| GNAQ | 3 | 78 | 0.089 | 0.097 | 3.749 | 0.818 ~ 17.190 |
| NKX2_1 | 3 | 78 | 0.098 | 0.098 | 3.615 | 0.790 ~ 16.551 |
| CCNE1 | 5 | 76 | 0.097 | 0.098 | 2.572 | 0.843 ~ 0.098 |
Table 3
The association between the mutated genes and progression-free survival.
| Gene | Case number with mutation | Case number without mutation | p value | Adjusted p value | Hazard ratio | 95% interval (Lower ~ Upper) |
| MET | 24 | 57 | 0.044 | 0.067 | 2.081 | 1.020 ~ 4.248 |
| BCL9 | 4 | 77 | 0.059 | 0.067 | 3.274 | 0.958 ~ 11.194 |
| TSC2 | 27 | 54 | 0.029 | 0.067 | 2.122 | 1.079 ~ 4.177 |
| NF1 | 48 | 33 | 0.052 | 0.067 | 2.222 | 0.993 ~ 4.974 |
| MAP2K2 | 2 | 79 | 0.063 | 0.067 | 4.066 | 0.926 ~ 17.854 |
| NFE2L2 | 6 | 75 | 0.062 | 0.067 | 0.244 | 0.056 ~ 1.073 |
| U2AF1 | 3 | 78 | 0.008 | 0.067 | 5.395 | 1.563 ~ 18.615 |
| CHEK2 | 4 | 77 | 0.049 | 0.067 | 3.503 | 1.007 ~ 12.184 |
| VHL | 8 | 73 | 0.020 | 0.067 | 3.293 | 1.206 ~ 8.993 |
| CBL | 2 | 79 | 0.016 | 0.067 | 6.589 | 1.425 ~ 30.475 |
| IFITM1 | 79 | 2 | 0.023 | 0.067 | 0.168 | 0.036 ~ 0.778 |
| IDH2 | 2 | 79 | 0.049 | 0.067 | 4.398 | 1.005 ~ 19.250 |
| DNMT3A | 9 | 72 | 0.036 | 0.067 | 0.267 | 0.078 ~ 0.918 |
| BTK | 2 | 79 | 0.016 | 0.067 | 6.589 | 1.425 ~ 30.475 |
| PPP2R1A | 8 | 73 | 0.087 | 0.087 | 2.329 | 0.883 ~ 6.143 |
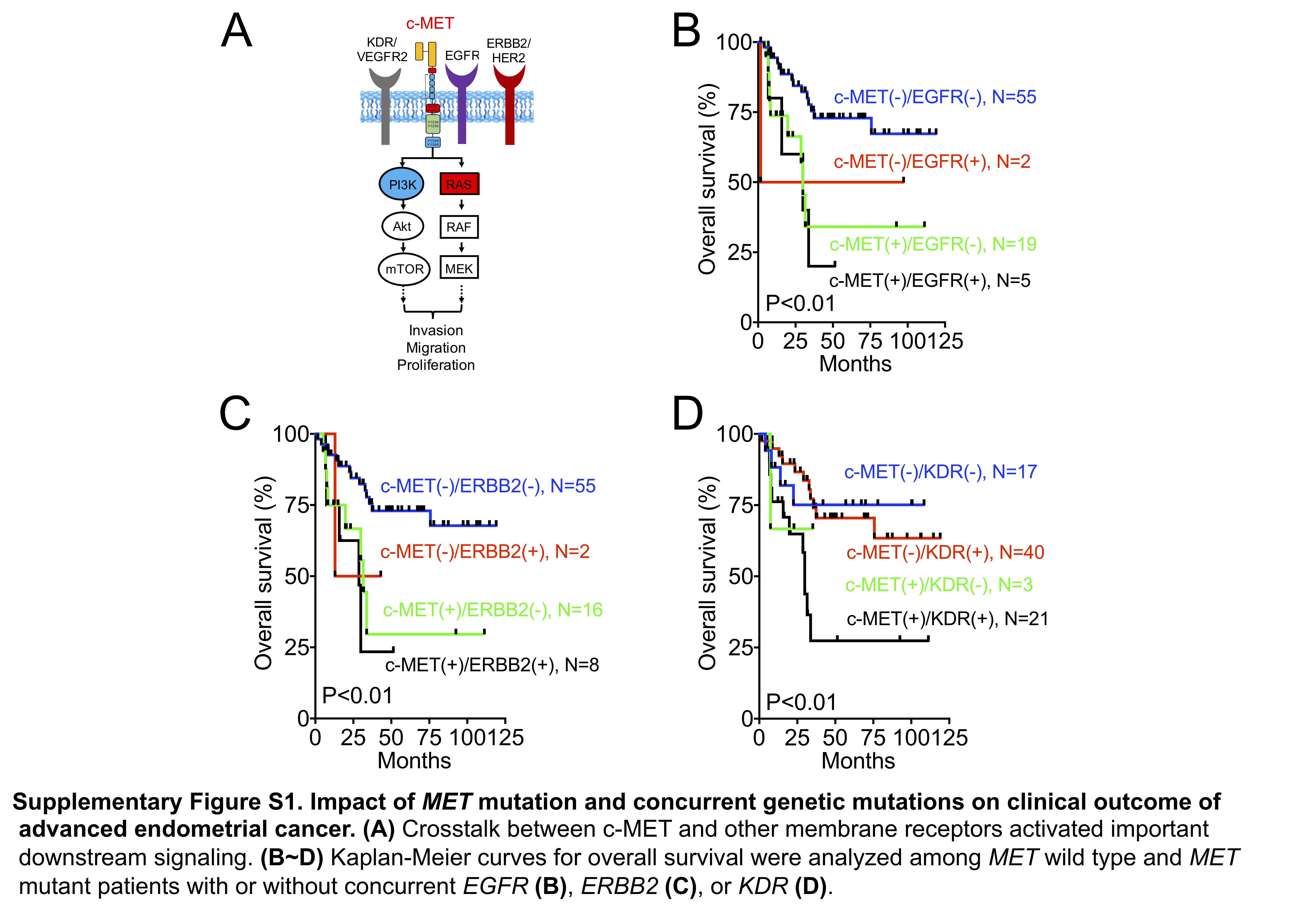
MET mutation is a cancer driver. MET mutation is a poor clinical marker (Fig. 1A). c-MET, a protein encoded by the human MET gene, is a receptor tyrosine kinase (RTK) expressed on the cell surface (1). The aberrant activation of the c-MET pathway and crosstalk with other RTKs has been shown to stimulate the PI3K/AKT and RAS/MAPK signaling pathways, which contribute to cancer biology (Supplementary Fig. 1A). MET mutation was associated with poor survival independent of EGFR mutation (Supplementary Fig. 1B). Alternatively, EGFR mutation did not affect the clinical outcome in patients harboring the wild-type or mutant MET. Similar results were observed in the MET and ERBB2, MET and PIK3R1, and MET and PIK3CA combination mutations (Supplementary Fig. 1C, Figs. 1B and 1C). However, the impacts of KDR and KRAS mutations were different. The presence of KDR or KRAS mutations indicated poor outcomes in patients with MET mutations, but not in those without MET mutations (Supplementary Fig. 1D and Fig. 1D). Taken together, MET mutation highly influences the clinical outcome of advanced endometrial cancer, and KDR and KRAS mutations exhibit additional impacts on patients with MET mutation.
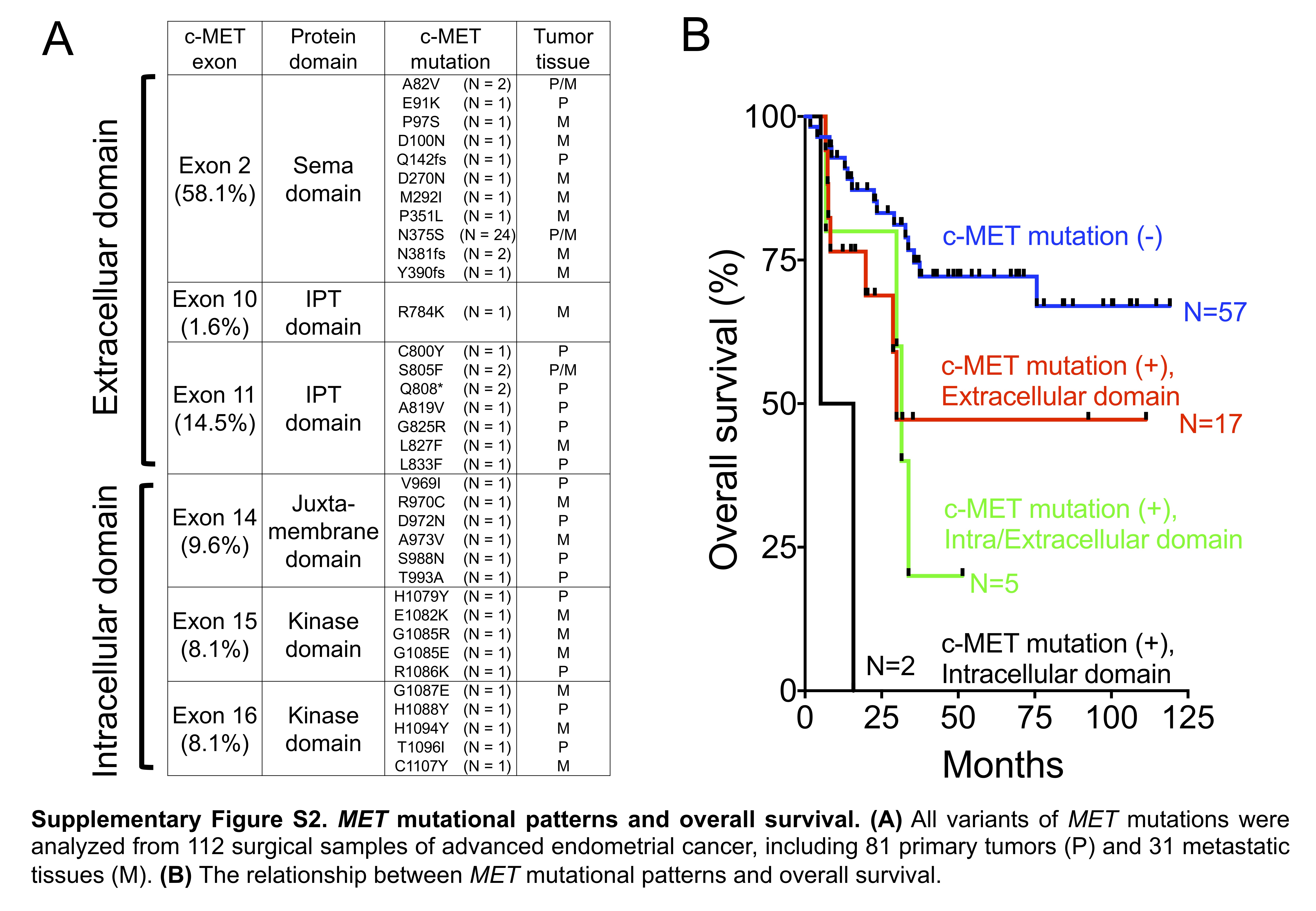
In silico analysis of MET mutation. The types of MET mutations were analyzed in 112 samples, including 81 primary tumors and 31 metastatic tissues. A total of 35 nonsynonymous mutations, including missense mutations and small INDELs, were identified in exons 2, 10, 11, 14, 15, and 16 (Fig. 2A, Supplementary Figure S2A). Aberrant MET mutations resulted in c-MET overexpression in the endometrial cancer tissues (Fig. 2B), similar to that seen in lung cancer, indicating that c-MET overexpression alone can induce oncogenic transformation in vitro and in vivo. 16,17,18,19 Moreover, patients with intracellular domain mutations seemed to exhibit a worse outcome (Supplementary Figure S2B).
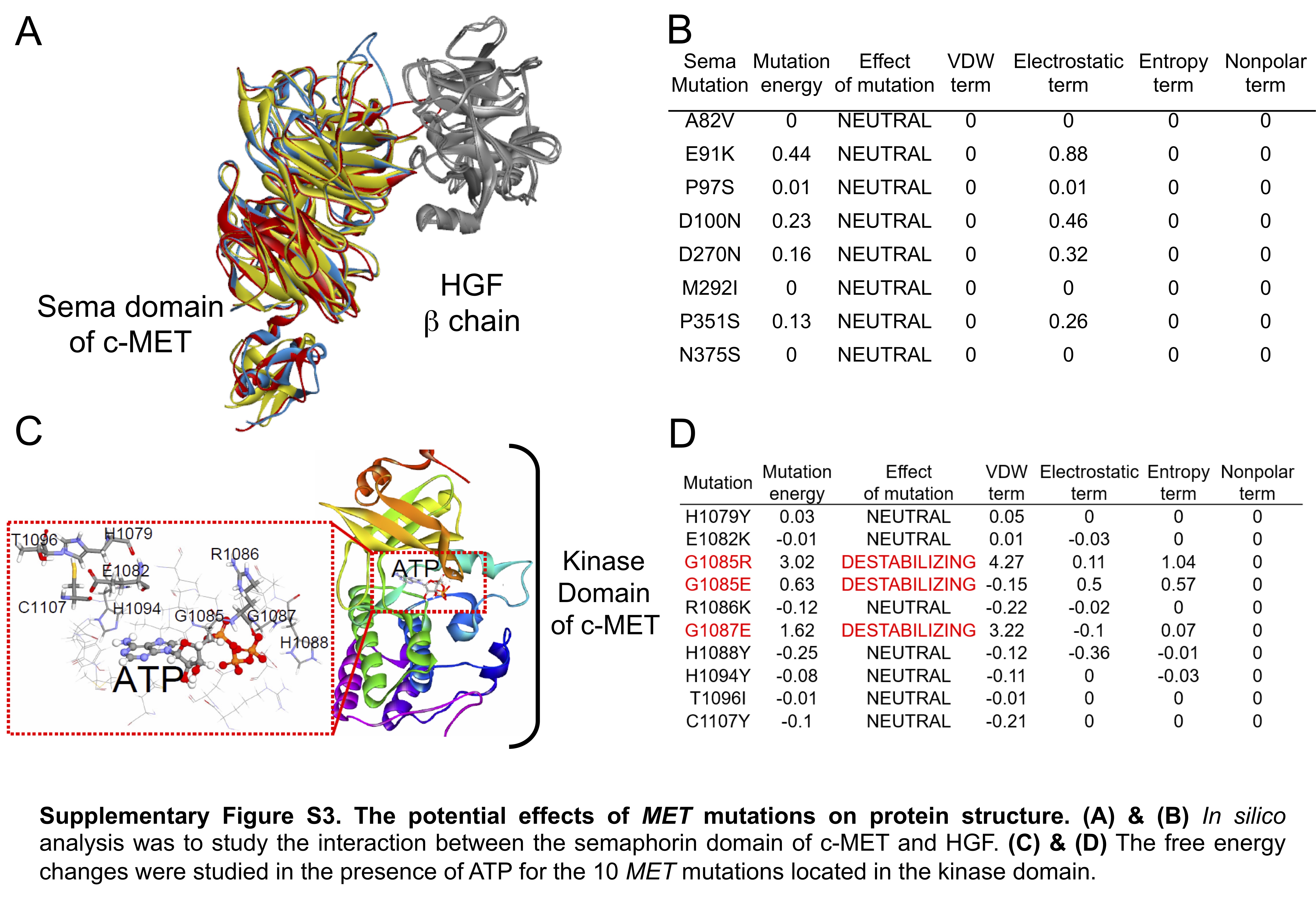
To predict the impact of the MET mutations on the protein structure, the “in silico” analysis was performed.20 When the interaction between the semaphorin domain of c-MET and HGF was analyzed (Supplementary Figure S3A), the exon 2 mutations in did not cause any obvious change in the predicted free energy, implying that these mutations did not theoretically affect the binding of HGF to c-MET (Supplementary Figure S3B). For mutations in the kinase domain, the free energy changes were studied in the presence of ATP (Supplementary Figure S3C). As shown in Supplementary Figure S3D, the “in silico” models demonstrated a Gly-to-Arg or Gly-to-Glu change at codon 1085 or a Gly-to-Glu change at codon 1087, which increased the mutational free energy. These findings indicated that these mutations might affect ATP binding, c-MET phosphorylation, and subsequent biological functions.
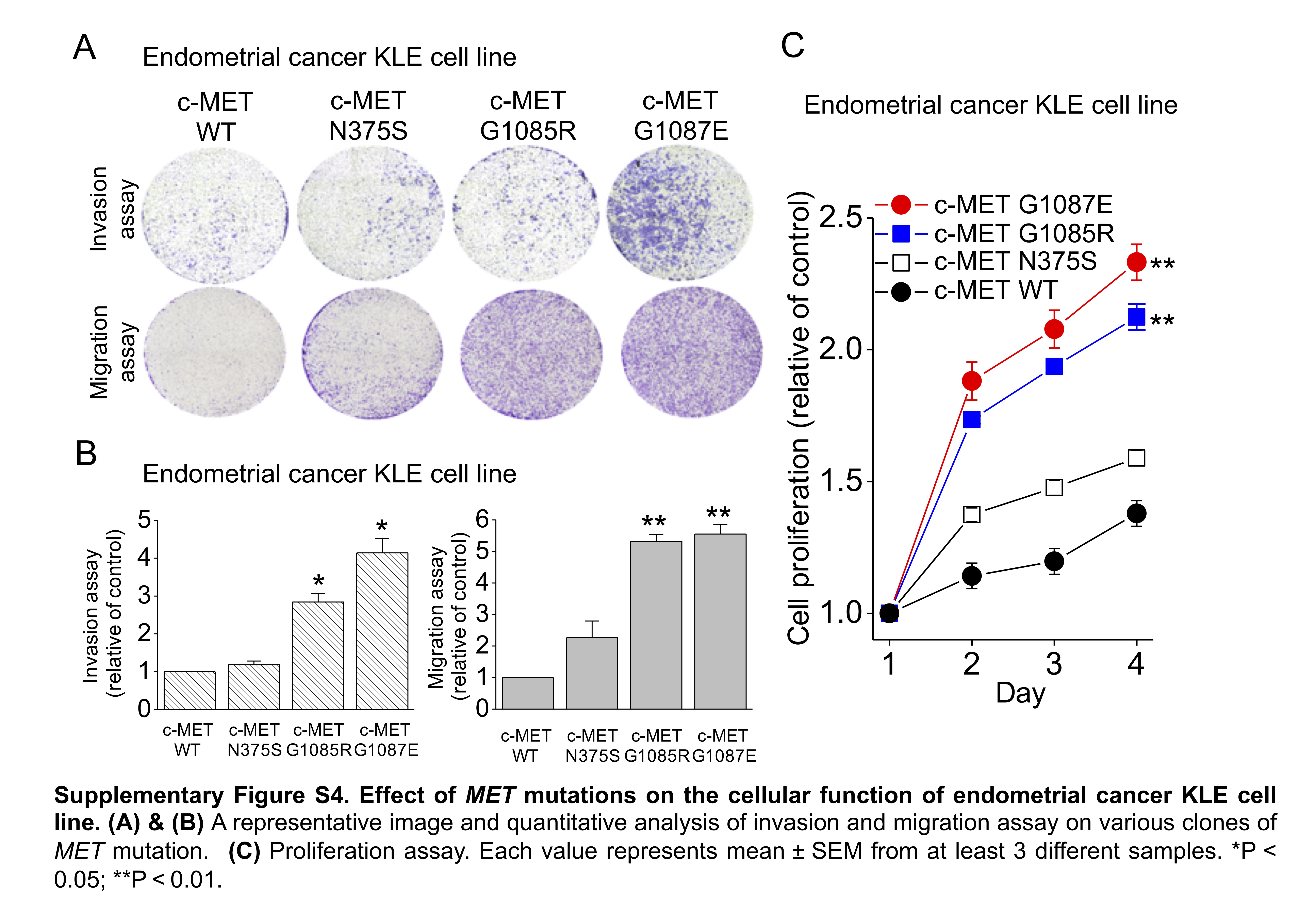
Effect of MET mutation on cellular function. To confirm the “in silico” analysis, we established various clones of MET mutants in the RL95-2 and KLE cell lines. The time course of c-MET phosphorylation was studied in response to HGF stimulation. In the wild-type c-MET RL95-2 cells, HGF induced a rapid increase in c-MET phosphorylation within 3 min, which was gradually decreased over 120 min (Fig. 2C). In contrast, an equivalent increase in c-MET phosphorylation was observed, which was sustained for 120 min in exons 15 (G1085E) or 16 (G1087E) in the clones of the kinase domain mutants. In the mutated semaphorin domain N375S clones, a delayed peak in c-MET phosphorylation was noted at 30 min, followed by rapid dephosphorylation. These results imply that different MET aberrations might exert differential effects on cellular functions. This hypothesis was confirmed by the proliferation, migration, and invasion assays in the endometrial cancer cell lines (Fig. 2D & 2E, Supplementary Figure S4).
Endometrial cancer growth in vivo. To test whether the c-MET signaling pathway was a therapeutic target, we subcutaneously inoculated the SCID mice with RL95-2 cells carrying various MET mutations. Tumors harboring different MET mutants showed a significant increase in volume when compared to those with wild-type MET (Fig. 3A-B, Supplementary Figure S5). Next, cisplatin significantly inhibited the growth of tumors carrying the wild-type MET and the G1085R and G1087E mutations (Fig. 3C-D). However, it appeared to exhibit no inhibitory effect on tumors with MET N375S. Crizotinib, a multi-target tyrosine kinase inhibitor, significantly inhibited the growth of tumors carrying the N375S, G1085R or G1087E mutant, when compared to those with the wild-type MET (Fig. 3E-F). SU11274, a specific c-MET inhibitor, also showed potent inhibitory effects on the growth of tumors carrying G1085R or 1087E mutations, whereas its inhibitory effect on the growth of tumors carrying wild-type MET was not very obvious. Interestingly, tumors with N375S mutation were insensitive to SU11274 in vivo (Fig. 3G-H). These data indicate that MET mutations promote the growth of endometrial tumors and show different sensitivities to cisplatin or tyrosine kinase inhibitors (crizotinib and SU11274), depending on the type of MET mutation.
MET N375S is a germline variant. In animal studies, mutation of the semaphorin domain N375S displayed resistance to cisplatin, the major chemotherapeutic agent for endometrial cancer (Fig. 3C and D). In our cohort, the OS rate for patients without MET mutations was 75%; however, that value significantly decreased to 50% in patients with MET N375S (Fig. 4A, P = 0.043). Furthermore, an analysis of 35 paired tissues revealed that the MET N375S mutation was a germline variant (Fig. 4B), consistent with the findings of a previous study in lung cancer.21 When compared to other nonsynonymous variants, the incidence of the N375S variant (c.1124 A > G) was relatively high (11 ~ 17%) in the current study and the Taiwan Biobank (Fig. 4C). The worldwide distribution of genetic N375S variants (c.1124 A > G) was assessed in the data from the 1000 Genomes Project. The allele frequency of the MET N375S variant (c.1124 A > G) was 8.5% and 5.8% in all the patients enrolled in the present study and the Taiwan Biobank, respectively. A similar allele frequency (5%~8%) was observed in the population from South and East Asia (Fig. 4D). In contrast, less than 2% of the alternative allele frequency was observed in the European, American, and African populations.
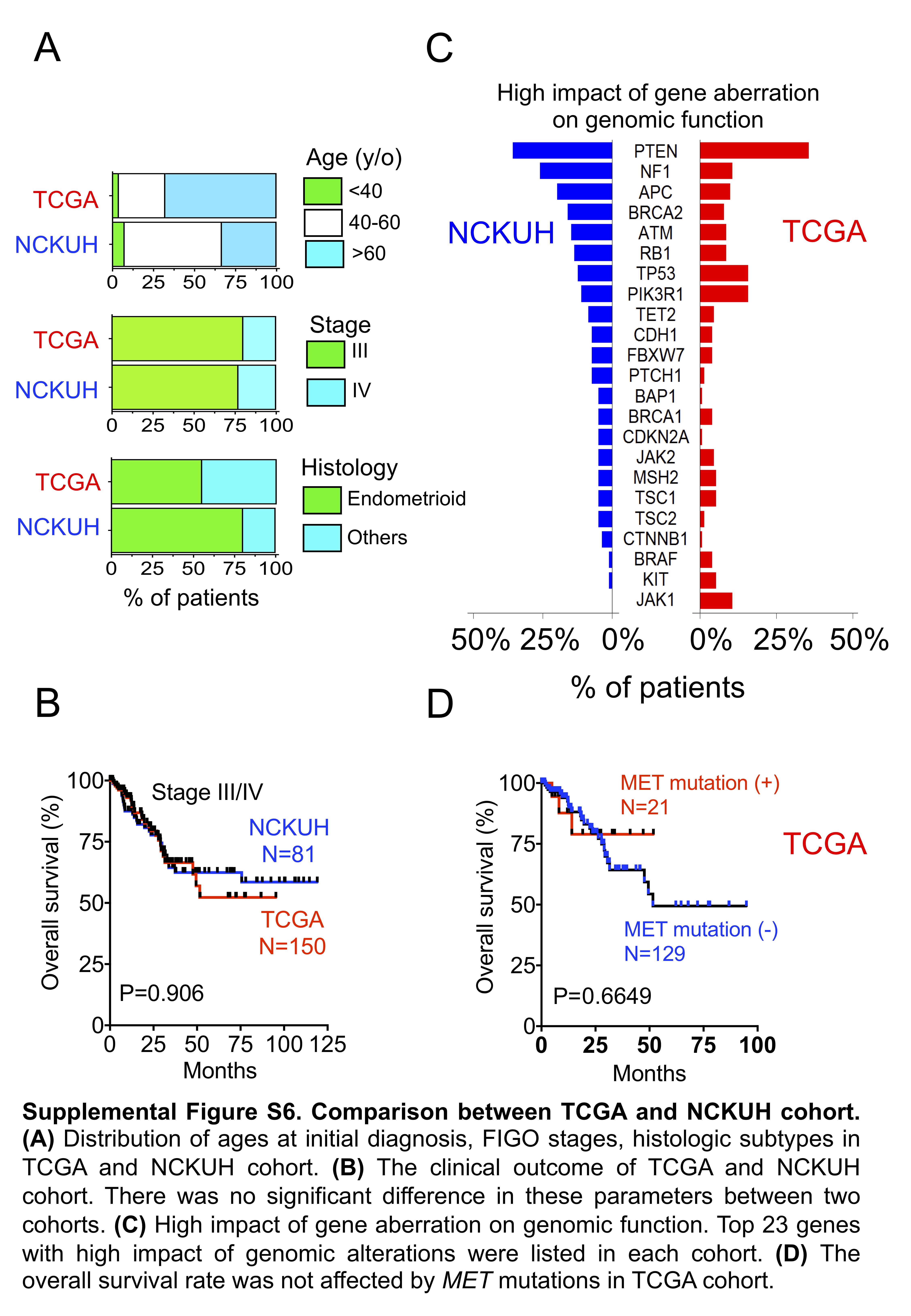
Comparison with TCGA molecular classification. Patients with advanced disease (stage III or IV) in the TCGA endometrial cancer cohort were identified, and the genomic backgrounds between the NCKUH and TCGA cohort were compared. No significant differences in age distribution at initial diagnosis, FIGO stage, histology, and clinical outcome were observed between the two cohorts (Supplementary Figure S6A and S6B). To obtain somatic mutations, the variants identified by Oncomine Comprehensive Assay v1 were filtered by the germline genetic variants. The top gene aberrations with high impact on genomic functions were listed after analyzing and filtering out the mutations, which were categorized as “low” (harmless or unlikely to change protein behavior), “modifier” (affecting the non-coding), or “moderate” (inframe insertion or protein-altering variant; Supplementary Figure S6C). The ranking of the top 10 genetic aberrations was different between the two cohorts, although PTEN was the most common mutated gene. Mutations in BRAF, JAK1, and KIT were identified in approximately 4–11% of the patients in TCGA cohort, but they were rarely detected in the NCKUH cohort. In addition, at the advanced stage, MET mutations were observed in 15% of cases in the TCGA cohort without an impact on the OS (Supplementary Figure S6D).
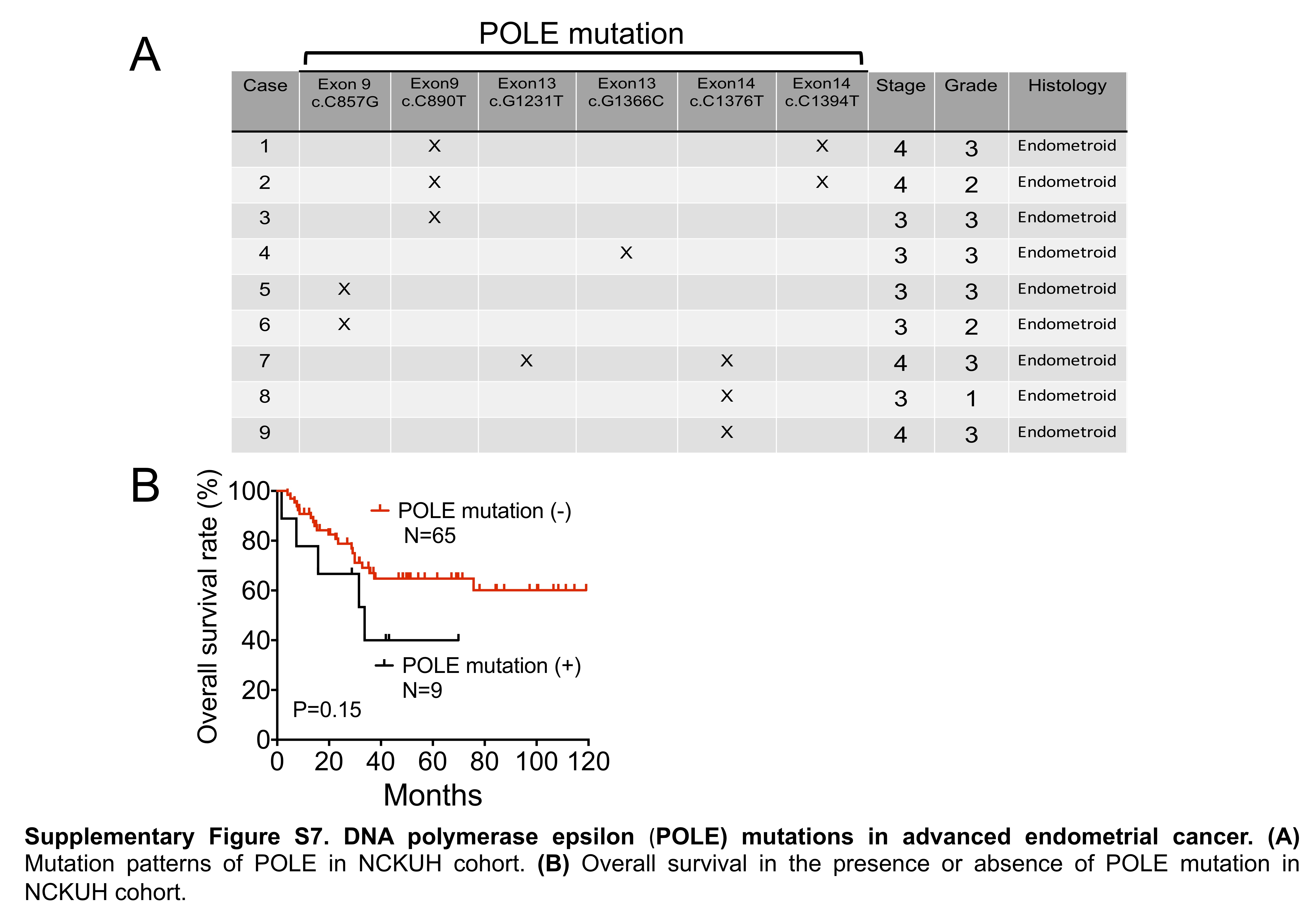
A subset of endometrioid cancer was newly identified in hotspot POLE mutations in the TCGA cohort.5 TCGA and subsequent studies showed that POLE-mutant endometrial cancers typically present as high-grade or poorly-differentiated tumors.5,22 In addition, POLE mutations accompanied by an ultra-tumor mutation burden present with favorable clinical outcomes.5,23 Among the 81 patients recruited in this study, 74 presented with adequate and qualified samples to investigate the pathogenic mutations in the POLE gene. However, no association between the POLE mutation and the histology of the specimens was observed in the current study cohort (Supplementary Figure S7A). In addition, the OS was not affected by the presence of the POLE mutation (Supplementary Figure S7B). A discrepancy in the genetic aberrations between the NCKUH and TCGA cohorts was noted.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}