Homozygous BCL10 mutation in a patient with combined immunodeficiency
We investigated a female patient coming from a consanguineous marriage from Saudi Arabia. She presented at one year of age with fever and bacterial pneumonia that required hospitalization. Her immune workup was suggesting of hypogammaglobulinemia and lymphocytosis (see supplementary material and Table D1 for detailed clinical history). Furthermore, she had a sister who died at 12 months of age due to a severe chest infection. These observations were compatible with a PID, and she underwent WES. WES revealed a homozygous nonsense mutation (A/T) affecting the nucleotide position g.85270779 (GRCh38.p12) of exon 2 of the gene encoding BCL10 in genomic DNA (gDNA) extracted from leukocytes (g. 85270779A > T). This mutation affects the lysine at position 63 and generates a premature stop codon (c.187A > T, p.K63X). The mutation K63X has not been reported in public databases such as ExAC or gnomAD, suggesting that it is private for this kindred. All other family members were healthy and heterozygous for the mutation (Fig. 1A and 1B). We then assessed BCL10 expression in PBMC from the patient, healthy controls, and heterozygous carriers. No BCL10 protein was detected on PBMCs from the patient, but BCL10 was detectable in the heterozygous carrier and a healthy donor (Fig. 1C). Our results indicated that this patient has a BCL10 complete deficiency.
Overt immunological phenotype in BCL10 deficient patient
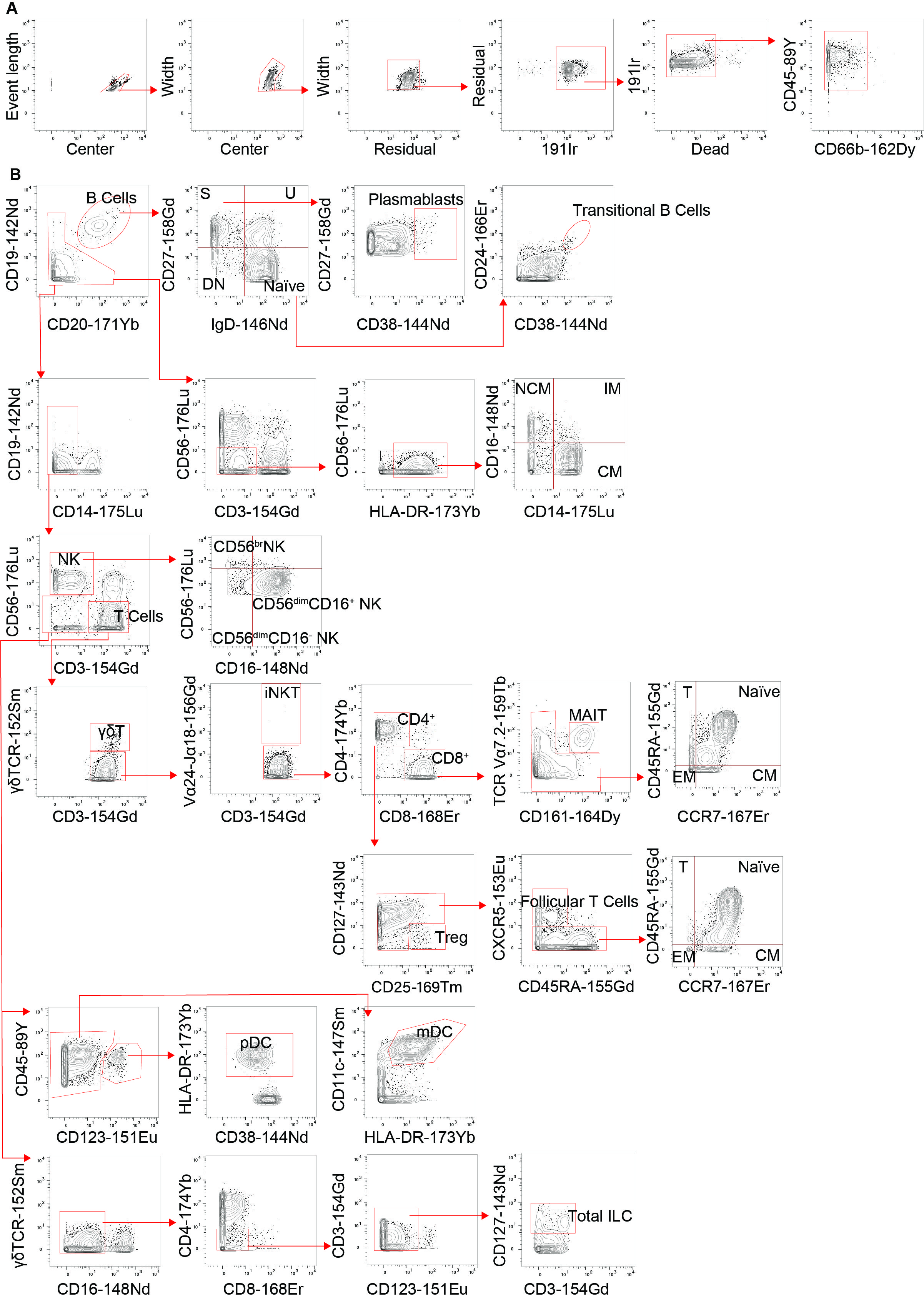
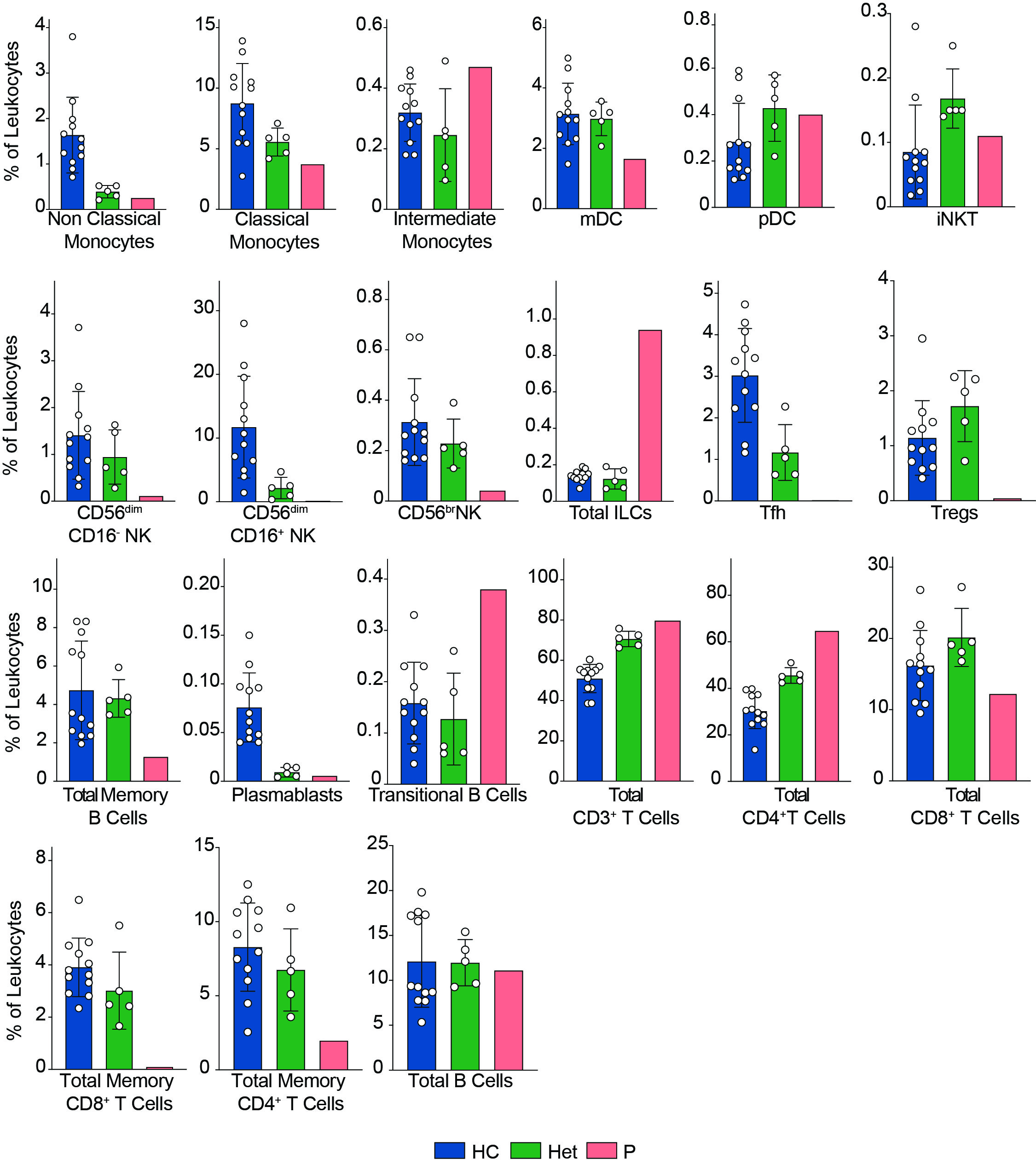
We previously reported that human BCL10 is critical for the proper development and function of hematopoietic and non-hematopoietic lineages[29]. The paucity of patients with BCL10 deficiency and the limited amount of samples obtained from these patients due to the severity of their disease hindered our capacity to survey the consequences of the absence of BCL10 in the overall composition of circulating leukocytes. Furthermore, the low number of individuals reported with deleterious heterozygous variants in BCL10 prevented us from assessing the consequences of reduced dosages of BCL10 in leukocyte development and function. The samples obtained from P3, her five healthy heterozygous carrier relatives, and the advent of mass cytometry provided the tools to tackle these issues. We performed in-depth immunophenotyping of the patient, healthy heterozygous carriers, and healthy controls using mass cytometry and a cocktail of 33 antibodies directed against surface markers designed to identify most of the common leukocyte populations as well as some rare ones (Table S2). Visualization using dimensional reduction with the t-SNE algorithm revealed marked differences in the distribution of leukocyte populations when comparing a healthy control and heterozygote carrier with the patient (Fig. 2A-C). Of the subpopulations identified by unsupervised clustering followed by manual clustering (Fig. 2A-C), mucosal-associated invariant T cells (MAIT), γδT and natural killer (NK) cells showed a reduction in the patient when compared with healthy controls and heterozygous carriers (Fig. 2D). This reduction in leukocyte subpopulations is further confirmed by manual gating (Fig. 2E and S2). Considering that the patient was 1.5 years of age at the time of blood draw and the frequencies of MAIT cells at that age range from 0 to 3% of PBMCs, we cannot conclude that the reduction observed in the patient in BCL10 dependent [34]. These results suggest that BCL10 deficiency causes changes in the leukocyte population distribution otherwise not seen in healthy individuals. Despite the frequencies of CD4+, CD8+, B cells, and myeloid cells in the patients fall within normal ranges when compared with healthy controls (Fig. 2D), the distribution of cells within each of these populations differs between groups suggesting a subtler difference in their subpopulations (Fig. 2A, B). To further characterize these differences, we reclustered each of these populations and performed an unbiased computational analysis.
BCL10 impairs memory B cell differentiation
BCL10 is located downstream of the B-cell receptor (BCR). Upon BCR activation, the LYN kinase phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMS). These events start a molecular cascade that culminates in the activation of PI3K and a phospholipase called PLCγ2. This molecule mediates the formation of diglycerol (DAG) as a second messenger to activate protein kinase C, which will act on CARD11 and promote the formation of the CBM complex. The formation of the CBM complex leads to the activation of the IκB complex kinase (IKK) complex, which will result in the activation of NF-κB [2]. Unsupervised clustering in the B cells from Fig. 2 identified two major populations (Fig. 3A). The frequency of cluster 2 in the patient was severely reduced compared to healthy controls and heterozygous carriers (Fig. 3B). We used marker enrichment modelling (MEM) to study these two clusters [33]. By using machine learning, MEM identifies the markers that distinguish each population allowing for an unbiased characterization of populations in an unbiased and automatic fashion. This analysis showed that cluster 1 is characterized by the expression of IgD and CD38 and cluster 2 by CD27, CD24, and CD25 (Fig. 3C). These markers are consistent with cluster 1 corresponding to naïve B cells and cluster 2 corresponding to memory B cells [35, 36]. We confirmed this observation by manual gating and observed that the patient has barely any detectable double negative or switched memory B cells with the naïve and unswitched compartments been comparable with the healthy control and heterozygous carriers (Fig. 3D, E). Our findings and the apparently abolished class switch observed in this, and the previous patients suggest that BCR signalling is dependent on BCL10. Hence, these results show that BCL10 is essential for naïve B cells to differentiate into their respective memory subpopulations.
Reduction of memory T cells in the absence of BCL10
We and others have shown that BCL10 is critical for TCR-mediated T cell activation in humans [29]. Upon TCR activation, lymphocyte-specific tyrosine kinase phosphorylates ITAMs leading to the activation of PI3K and the phospholipase PLCγ1. As is the case for B cells, these signalling events culminate in the formation of the CBM complex, which will end in the translocation of NF-κB into the nucleus and promote cell proliferation. We studied the consequence of the defective TCR activation caused by the absence of BCL10 in the composition of the T cell compartment [2]. We subsetted the CD4+ and CD8+ T cells identified in Fig. 2 and analysed them individually. By unsupervised clustering of the CD4+ cells, we observed three major cell populations (Fig. 4A). Population 2 and 3 were severely reduced in the patient compared to healthy controls and heterozygous carriers (Fig. 4B). MEM analysis showed that cluster 1 is characterized by CD27, CCR7, CD45A, and CD38 while cells from cluster 2 express high levels of CD27, CD45RO, CCR7, CXCR3, and CD25, and cluster 3 is characterized by CD45RO expression. This differential marker expression is compatible with cluster 1 corresponding to naïve CD4+ T cells and clusters 3 and 4 with memory CD4+ T cells (Fig. 4C). We studied the CD4+ memory and naïve compartments by manual gating (Fig. 4D, E). This analysis showed reduced central memory (CM), effector memory (EM), and TEMRA CD4+ T cell compartments with an increased naïve CD4+ T cell compartment in the patient when compared to the healthy controls or heterozygous carriers. Furthermore, by manual gating, we also observed a reduction in the frequency of Tregs and TFH in the patient (Fig. S2). These results suggest that BCL10 is necessary for the differentiation from naïve to memory in CD4+ T cells.
Similarly, unsupervised clustering of the CD8+ T cell population rendered three major clusters, two of which were almost absent in the patient compared to the healthy controls and heterozygous carriers (Fig. 5A, B). Marker characterization by MEM showed that cluster 1 expressed markers characteristic of naïve CD8+ T cells such as CD45RA, CCR7. In contrast, clusters 2 and 3 are characterized by the expression of the central memory and effector memory markers (CD45RO, CD127) [37] and or TEMRA markers (CD45RA), respectively (Fig. 5C). We studied the naïve and memory CD8+ T cell compartment by manual gating. Similarly to our observations in CD4+ T cells, we observed an increased naïve CD8+ T cell population in the patient compared to the healthy controls or heterozygous carriers. It also revealed decreased levels of central memory, effector memory, and TEMRA CD8+ T cells (Figs. 5D, E). The absence of these populations in the patient suggests that BCL10 is necessary for developing the memory CD8+ T cells compartment and confirms our previous results [29, 30]. As in B cell analysis, we did not observe any difference in the samples from heterozygous carriers, suggesting that haploinsufficiency for BCL10 is immunologically silent.
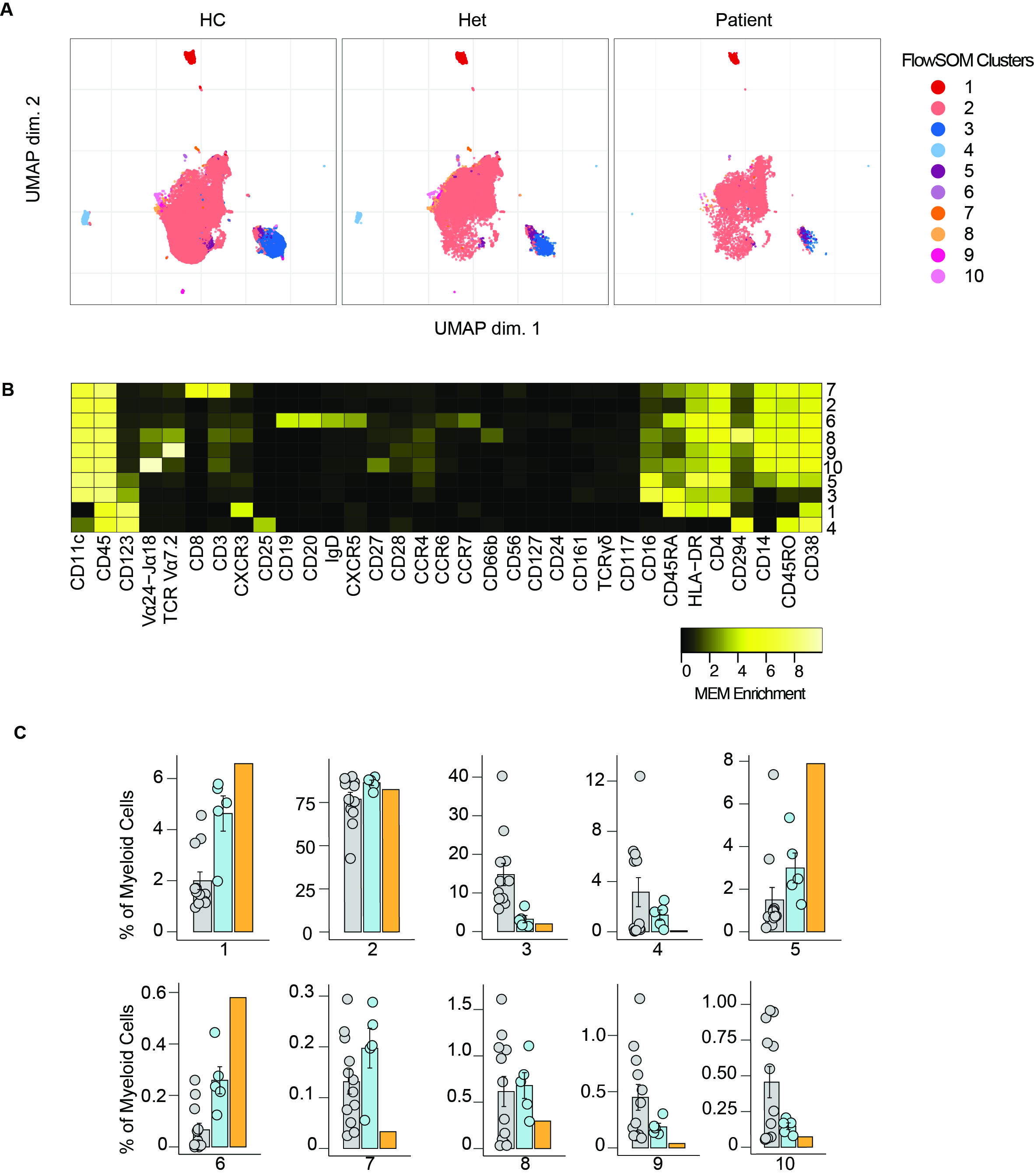
Normal myeloid compartment in BCL10 deficiency
Mice studies show that BCL10 is critical for NF-κB activation in myeloid cells. This is mediated by the TLR4, TLR2, and TRL6 pathways, which are BCL10 dependent. Our previous results showed that in humans, BCL10 is redundant for NF-κB activation in these cells [29]. Nevertheless, we performed unsupervised clustering in the myeloid cluster from Fig. 2. We studied 10 clusters and observed that the frequencies of these clusters were comparable between healthy controls and heterozygous carriers with the patient (Fig. S3). By manual gating, we showed that the frequencies of non-classical monocytes, classical monocytes, intermediate monocytes, myeloid dendritic cells, and plasmacytoid dendritic cells were comparable between healthy controls, heterozygous carriers, and the patient confirming the redundant role of human BCL10 in the development of cells from the myeloid lineage.
Clinical features in BCL10 deficiency
The patient presented in this study had a clinical presentation consistent with the previous two patients [29, 30] with respect to her bacterial lung infection (supplementary note 1: case report). She had a sister who died at the age of 15 mo due to disseminated BCGitis and bacterial sepsis. She did not develop any gastrointestinal manifestations, and her clinical course was cured by HSCT.
{kind=link}
{kind=link}
{kind=link}