In the present study, four novel quercetin derivatives were designed and synthesised by one pot synthesis method using benzoic acid and its derivatives. The synthesised compounds were screened for physicochemical and drug-likeness properties, evaluated for in vitro antioxidant assays such as hydrogen peroxide (H2O2) and 2,2-diphenyl-1-picryl hydrazyl (DPPH), cytotoxicity study on breast cancer cell line MCF-7 and performed molecular docking against the nitric oxide synthase (iNOS) enzyme 4NOS PDB (Protein Data Bank) ID which is expressed in breast cancer. In the screening of physicochemical and drug-likeness properties, QB, QB1 and QB4 are eligible for oral drug screening and other derivatives QB2 and QB3 were not eligible for oral drug screening. Among all, QB-1 showed highest percentage of inhibition and lowest IC50 value in both the assays. The docking results displayed that QB-2 showed highest docking score and exhibited highest cytotoxicity against MCF-7 cell line and the study results conclude that QB1 and QB2 compounds can be further explored to in vivo anticancer activity.
Research Article
Design, Synthesis, Physicochemical, Drug-likeness Properties of Quercetin Derivatives and Their Effect On MCF-7 Cell Line and Free Radicals
https://doi.org/10.21203/rs.3.rs-817941/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Quercetin derivatives
drug-likeness
physicochemical properties
antioxidant activity
cytotoxicity
molecular docking
The generation of free radicals is a key factor that can lead to development of various diseases related to central nervous system, cardiovascular system, respiratory, skeletal, excretory systems, metabolism and multi-organs. These free radicals are either produced endogenously due to inflammation, stress, immune cell activation, ischemia, exercise, infection, aging, cancer or through exogenous substances such as cigarette smoking, alcohol consumption, heavy and transition metals, pharmaceutical agents, radiation and air and water pollutants.1 Flavonols, the largest subclass of flavonoids is widely found in fruits and vegetables such as grapes, berries, apples, broccoli, onions and citrus fruits. They are characterised by the presence of a ketone group at C4 along with an unsaturation between the C2 and C3 position and hydroxyl group at C3 in the C ring. Quercetin is one of the most common flavonol present in several plant foods. Structurally, quercetin is 3,5,7,3’,4’–pentahydroxyflavone (2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one) and the presence of five hydroxyl groups is responsible for its versatile biological activities such as antioxidant, anticancer, antimicrobial, anti-inflammatory and for treating age-related, cardiovascular and neurodegenerative disorders (Figure 1).2,3
Quercetin is capable of exhibiting radical scavenging activity for protection of body cells against free radicals that are produced during metabolism process or from external sources such as pollution, cigarette smoke, radiation and medication. Perhaps, according to the studies, quercetin faces limitation about its bioavailability due to poor absorption of quercetin because of low solubility of quercetin and the first-pass effect of quercetin in the liver and gut.4,5 Studies have shown that the introduction of ester group into the quercetin scaffold has produced potent compounds for antioxidant, anticancer (anti-proliferative) and also enhance the bioavailability as well.6 Danihelová et al., 2013 prepared fifteen quercetin-derived compounds by condensation or selective protection reactions followed by acyl chloride acylation, which showed strong cytotoxicity towards HeLa cells.7 Other quercetin derivatives synthesized by esterification with aspirin at the 3- and 7-OH groups reported for higher cytotoxic effects against liver (HepG2) and promyelocytic leukemia (HL-60) cancer cells8 and also anti-hepatitis C virus activity.9 Hence, chemical modification of quercetin can generate novel derivatives with improved biological effects, bioavailability and solubility properties. Therefore, in the present study we aimed for one-pot synthesis of quercetin esters, physicochemical and drug-likeness properties, molecular docking and its biological evaluation.
Chemistry
All the chemicals, reagents and solvents used for experimental work were analytical grade. The formation of product was monitored by performing TLC (using Silica gel F254 Aluminium sheets from Merck Company). Column chromatography was carried out by using Silica gel 100-200 mesh. Open capillary method was performed to determine the melting point of the synthesised quercetin derivatives and was expressed in units of ºC. IR spectral study was carried out using Shimadzu FTIR 8400-S spectrophotometer by KBr Press method and IR data obtained was expressed in cm-1. 1H NMR and 13C NMR studies were carried out on Bruker 400 MHz FT-NMR spectrophotometer using trimethylsilane as internal standard and DMSO (D6) and CDCl3 as solvent systems. Mass spectra were recorded on LC-MS ACQUITY UPLC mass spectrometer using AP and ESI method and TOF detector.
Synthesis of quercetin derivatives
Synthesis of 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-3-yl benzoate (QB1)
Quercetin dihydrate (2 mmol), benzoic acid (6 mmol) and zinc chloride (2 mmol) were taken in a RBF. 10 ml of phosphorous oxychloride was gradually added to the mixture and was stirred for 15 hrs at 75 °C. The reaction completion was supervised by TLC. Upon formation of the product, stopped stirring and the product was allowed to cool. Ice cold water was added and then product was extracted using ethyl acetate.10 The separated ethyl acetate layer was collected and evaporated. The obtained brownish yellow product was subjected to preparative TLC, using chloroform: methanol (70:30) as the mobile phase. The pure product was separated, collected and dried. Yield: 80 mg (10%); m.p. 220-223 ˚C; FT-IR (cm-1): 3360.11 (alcohol O–H stretch), 3070.78 (Ar – H), 1695.49 (C=O stretch), 1506.46 (C=C stretch), 1174.59 (C–O ester);1H NMR (400 MHz [D6]DMSO): δ 6.18 (s, 1H, ArH(6)), 6.45 (s, 1H, ArH(8)), 7.13 (d, J = 6.4 Hz, 1H, ArH(14)), 7.47 (s, 1H, ArH(11)), 7.59 (t, J = 2.4 Hz, 2H, ArH(19)), 7.74 (d, J = 5.6 Hz, 1H, ArH(15)), 7.93 (t, J = 8.4 Hz, 1H, ArH(20)), 8.13 (d, J = 3.2 Hz, 2H ArH(18)), 9.32 (s, 3H, OH(7,12,13)), 12.46 (s, 1H, OH(5)); 13C NMR (400 MHz [D6]DMSO): δ 93.97 (C8), 98.85 (C6), 104.89 (C4), 113.24 (C11), 116.15 (C14), 122.72 (C15), 124.32 (C10), 128.97 (C19), 129.39 (C18), 129.7 (C2), 130.1 (C20), 130.33 (C17), 131.4 (C12), 133.2 (C13), 155.81 (C1), 157.95 (C9), 161.12 (C5), 164.21 (C16), 164.53 (C7), 167.86 (C3); LC-MS, m/z: calculated for C22H14O8 is 406.07; found 407.01 [M+H] +.
Synthesis of 2-(3,4-dihydroxyphenyl)-7-hydroxy-4-oxo-4H- chromene-3,5-diyl bis(2-chlorobenzoate) (QB2)
Quercetin dihydrate (2 mmol), 2-chlorobenzoic acid (5 mmol) and zinc chloride (2 mmol) were taken in a round bottom flask (RBF). Then, 10 ml of phosphorous oxychloride was gradually added to the reaction mixture and stirred for 15 hrs at 75 °C. The reaction completion was supervised by thin layer chromatography (TLC). Upon formation of the product, stopped stirring and the product was allowed to cool. Ice cold water was added to the mixture and product was extracted using ethyl acetate. The separated ethyl acetate layer was collected and evaporated.10 The obtained yellow product was subjected to preparative TLC, using chloroform: methanol (60:40) as the mobile phase. The pure product was separated, collected and dried. Yield: 100 mg (8.7%); m.p. 230-232 °C; FT-IR (cm-1): 3344.68 (alcohol O–H stretch), 2924.18 (Ar – H), 1726.35 (C=O stretch), 1654.99 (C=C stretch), 1172.76 (C–O ester), 725.78 (C–Cl ); 1H NMR (400 MHz [D6]DMSO): δ 6.037 (s, 1H, ArH(8)), 6.179 (s, 1H, ArH(6)), 6.45 (s, 1H, ArH(11)), 6.72 (d, J = 4.4 Hz, 1H, ArH(14)), 7.20 (d, J = 3.6 Hz, 1H, ArH(15)), 7.69 (t, J = 8 Hz, 4H, ArH(20, 21)), 8.08 (d, J = 10.4 Hz, 2H ArH(19)), 8.23 (d, J = 2.4 Hz, 2H ArH(22)), 9.375 (s, 3H, OH(7,12,13)); 13C NMR (400 MHz [D6]DMSO): δ 94.43 (C8), 98.95 (C6), 102.12 (C4), 114.97 (C14), 115.08 (C11), 119.87 (C10), 121.27 (C15), 128.02 (C21), 129.41 (C17), 129.76 (C20), 130.87 (C19), 131.65 (C22), 133.59 (C18), 141.73 (C2), 144.58 (C12), 144.67 (C13), 147.58 (C5), 153.33 (C1), 153.42 (C9), 160.16 (C7), 166.21 (C16), 174.03 (C3); LC-MS, m/z:exact mass calculated for C29H16Cl2O9 is 578.02; found 578.85.
Synthesis of 2-(3,4-dihydroxyphenyl)-7-hydroxy-4-oxo-4H-chromene-3,5-diyl bis(4-chlorobenzoate) (QB3)
The quercetin dihydrate (3 mmol), 4-chlorobenzoic acid (8 mmol) and zinc chloride (3 mmol) were transferred in a RBF. Then 10 ml of phosphorous oxychloride was gradually added and the reaction mixture was stirred for 15 hrs at 75 °C. The reaction completion was monitored by TLC and upon formation of the product the reaction was stopped and mixture was allowed to cool. Ice cold water was added and then extracted using ethyl acetate. The separated ethyl acetate layer was collected and evaporated. The obtained pale yellow product was subjected to column chromatography using dichloromethane: ethyl acetate (50:50)) as the mobile phase. The pure product was separated, collected, and dried. Yield: 154 mg (9%); m.p. 240-242˚C; FT-IR (cm-1): 3363.97 (alcohol O–H stretch), 2924.18 (Ar – H), 1743.71 (C=O stretch), 1687.77 (C=C stretch), 1139.97 (C–O ester), 725.82 (C–Cl ); 1H NMR (400 MHz CDCl3): δ 6.07 (s, 1H, ArH(8)), 6.25 (s, 1H, ArH(6)), 6.54 (s, 1H, ArH(11)), 6.85 (d, J = 5.6 Hz, 1H, ArH(14)), 7.25 (d, J = 4.8 Hz, 1H, ArH(15)), 7.51 (d, J = 2.4 Hz, 2H, ArH(24)), 7.74 (d, J = 2 Hz, 2H, ArH(19)), 8.18 (d, J = 4.8 Hz, 2H, ArH(23)), 8.35 (d, J = 4.8 Hz, 2H, ArH(18)), 9.672 (s, 3H, OH(7,12,13)); 13C NMR (400 MHz CDCl3): δ 94.2 (C8), 99.15 (C6), 103.95 (C4), 116.22 (C11), 116.23 (C14), 119.65 (C10), 121.99 (C15), 125.72 (C17), 126.41 (C22), 129.12 (C24), 129.24 (C19), 130.685 (C23), 130.72 (C18), 133.54 (C25), 133.76 (C20), 140.54 (C2), 144.81 (C12), 145.79 (C13), 148.47 (C5),153.34 (C1), 153.46 (C9), 161.1 (C7), 166.21 (C16), 166.32 (C23), 176.13 (C3); LC-MS, m/z: exact mass calculated for C29H16Cl2O9 is 578.02; found 578.92.
Synthesis of 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4-oxo-4H-chromen-3-yl 4-methyl benzoate (QB4)
Quercetin dihydrate (3 mmol), 4-methylbenzoic acid (8 mmol), zinc chloride (3 mmol) and 10 ml of phosphorous oxychloride was gradually added in the RBF. The reaction mixture was stirred for 15 hrs at 75 °C. The reaction completion was supervised by TLC. Upon formation of the product, stopped stirring and the product was allowed to cool. Ice cold water was added and to extract the product ethyl acetate was used. The separated ethyl acetate layer was collected and evaporated. The obtained orange-yellow product was subjected to column chromatography using dichloromethane: ethyl acetate (50:50)) as mobile phase. The pure product was separated, collected and dried.10 Yield: 127 mg (10.2%); m.p. 210-214 °C; FT-IR (cm-1): 3295.21 (alcohol O–H stretch), 2924.18 (Ar – H), 2860.53 (aliphatic C–H stretch), 1743.71 (C=O stretch), 1680.05 (C=C stretch), 1143.53 (C–O ester); 1H NMR (400 MHz CDCl3): δ 2.61 (s, 3H, ArH(21)) 6.34 (s, 1H, ArH(6)), 6.62 (s, 1H, ArH(8)), 7.694 (s, 1H, ArH(11)), 7.82 (d, J = 2 Hz, 1H, ArH(14)), 8.19 (d, J = 1.2 Hz, 1H, ArH(15)), 8.40 (d, J = 3.6 Hz, 2H, ArH(19)), 8.73 (d, J = 2.4 Hz, 2H, ArH(18)), 10.16 (s, 3H, OH(7,12,13)), 12.18 (s, 1H, OH(5)); 13C NMR (400 MHz CDCl3): δ 22.32 (C21), 91.48 (C8), 97.16 (C6), 103.76 (C4), 114.24 (C11), 117.39 (C14), 120.51 (C15), 122.25 (C10), 124.62 (C17), 129.79 (C19), 131.31 (C18), 133.42 (C2), 142.97 (C20), 143.84 (C12), 143.85 (C13), 149.17 (C1), 152.86 (C9), 160.93 (C5), 164.22 (C16), 165.49 (C7), 174.34 (C3); LC-MS, m/z: exact mass calculated for C23H16O8is 420.08; found 420.06.
Physicochemical, Pharmacokinetic and Drug-likeness Properties Screening and Prediction
The molecular descriptors, physicochemical properties and pharmacokinetic properties were evaluated using Swiss ADME web server (http://www.swissadme.ch/ accessed on 22 July 2021)11 and Pre-ADME (https://preadmet.bmdrc.kr/) were used to predict physicochemical, lipophilicity, water solubility, pharmacokinetic and drug-likeness properties including absorption, distribution, metabolism and excretion (ADME) and toxicity. The screening criteria of Drug-likeness compounds state that the synthesized compounds should be aqueous-soluble, should have high gastrointestinal absorption, orally active drug should satisfy Lipinski’s rule of five and low hERG inhibition risk (low toxicity).11
Biological Activity
In vitro antioxidant activity
The methods for in vitro antioxidant activity are based on the inhibition of free radicals. The inhibition of free radicals is measured when the samples are added to a system where free radicals are generated. The inhibition is relative to the antioxidant capacity of each sample. In vitro antioxidant methods provide an indication of antioxidant capacity of the compounds. In the present study, the synthesized quercetin derivatives were tested for in vitro antioxidant activity using DPPH and H2O2 assay.
Scavenging of H2O2 assay
A solution of H2O2 was prepared in phosphate buffer saline pH 7.4. Various concentrations (100-12.5 μg/ml) of samples and standards (ascorbic acid and quercetin) in methanol were prepared and added to H2O2 solution in PBS. After 10 min, the absorbance was measured at 230 nm.12,13
Scavenging of DPPH assay
The 100 μM of DPPH solution was prepared using methanol. The different concentrations of standard (ascorbic acid and quercetin) and quercetin derivatives in DMSO were prepared (100-12.5 μg/ml) and then added to DPPH solution. After 30 min, the absorbance for each was measured at 490 nm.12,13
In vitro cytotoxicity by MTT assay method
In vitro cytotoxicity was carried out in the Animal Tissue Culture Laboratory, Department of Pharmaceutical Biotechnology at JSS College of Pharmacy, Ootacamund, Tamilnadu, India. The in vitro cytotoxicity study involved the determination of mitochondrial synthesis by MTT assay and was performed on MCF-7 breast cancer cell line.
Preparation of test solutions
All the quercetin derivatives (QB1-4) were dissolved in distilled dimethyl sulphoxide (DMSO) separately and Dulbecco's Modified Eagle Medium (DMEM) was used to make volume with 2% inactivated fetal bovine serum (FBS) to obtain a stock solution of 1000 µg/ml concentration and sterilized by filtration and stored at ─20 °C till use. Serial dilutions were made to obtain lower dilutions.
Cell lines and culture medium
MCF-7 cell line was procured from National Centre for Cell Sciences (NCCS), Pune, India. For culturing cell lines, DMEM supplemented with 10% inactivated FBS, Penicillin (100 IU/ml), Amphotericin B (5 mg/ml) and Streptomycin (100 mg/ml) were used in 5% CO2 at 37 °C in a humidified atmosphere. The cells were dissociated with TPVG solution (0.2% trypsin, 0.02% Ethylene diamine tetraacetic acid, 0.05% glucose in phosphate buffer solution). The cultures were grown in 25 ml flat bottles and experiment was carried out in 96 well microtitre plates, considered as stock culture.
The monolayer cell culture was trypsinized and by using DMEM containing 10% FBS the cell count was adjusted to 1.0 x 105 cells/ml. The 0.1 ml of diluted cell line (approximately 10,000 cells) was added to a 96 well microtitre plate. The partial monolayer was formed after 24 hrs and the supernatant was flicked off. The monolayer was washed with DMEM medium. To the partial monolayer, 100 ml of different concentrations of synthesized compounds were added in microtitre plates. The plates were kept for incubation at 37 oC for 72 hrs in 5% CO2 atmosphere. The microscopic examination was carried out and in an every 24 hrs interval observations were made. The drug solutions in the wells were discarded after 72 hrs. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) in PBS (50 ml) was added to each well and gently shaken. The plates were again incubated for 3 hrs at 37 oC in 5% CO2 atmosphere. The supernatant was removed and 100 ml of propanol was added to the plates and gently shaken to solubilize the formed formazan. The plates were subjected for absorbance using a microplate reader at a wavelength of 540 nm. The percentage growth inhibition was calculated using the following formula and concentration of test drug needed to inhibit cell growth by 50% (CTC50) values was generated from the dose-response curve of selected cell line.14,15

Molecular Docking
The X-ray crystallographic structure of human iNOS protein with resolution of 2.25 Å was collected from RCSB Protein Data Bank (PDB ID: 4NOS) and selected as the target protein for docking of the four designed compounds and quercetin. All the ligands were sketched using ChemDraw Professional 15.0 software. Ligand and protein were prepared and docking was done through BIOVIA Discovery Studio 2019 software. Molecular docking was performed using CDOCKER algorithm which is a simulated annealing docking method that uses CHARMm force field based molecular dynamics to generate random conformations. Different conformations for ligand adopting rigid body rotations were generated and
–CDOCKER energy and interaction energies (kcal/mol) for the molecules were obtained.16
Chemistry
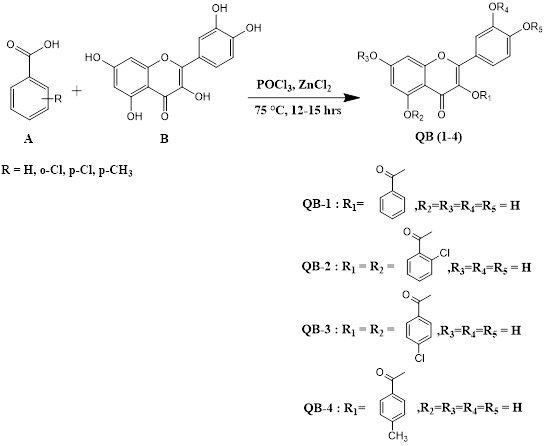
After thorough literature search and several trial and errors, we finally designed and carried out one-pot synthesis ester analogues of quercetin using benzoic acid and its derivatives namely o-chlorobenzoic acid, p-chlorobenzoic acid and p-methylbenzoic acid in the presence of phosphorusoxychloride and zinc chloride (Scheme 1). Compounds were obtained after purification by column chromatography and preparative TLC. The reaction of quercetin with benzoic acid in the presence of phosphorous oxychloride and zinc chloride catalyst afforded the quercetin-3-O-benzoic acid ester derivative (QB1). Purification was done by preparative TLC. The QB1 compound was obtained as a yellow solid (Table 1). The 1H NMR spectra of QB1 showed 10 proton peaks. Characteristic singlet signals at δ 6.18, 6.45, 7.47 ppm, doublet peaks at δ 7.74 and 8.13 ppm and triplet signals δ 7.59 and 7.93 ppm were observed and all corresponded to the aromatic protons, respectively. The two singlet signals at δ 9.32 and 12.46 ppm were assigned to the hydroxyl protons.
Treatment of quercetin dihydrate with o-chlorobenzoic acid in phosphorous oxychloride and catalysed by zinc chloride yielded quercetin-3,5-O-2-chlorobenzoic acid ester derivative (QB2). Compound was purified by preparative TLC. The QB2 compound was obtained as yellow solid. The 1H NMR spectra of QB2 showed nine proton peaks. It displayed characterised singlet peaks at δ 6.03, 6.17 and 6.46 ppm, doublet signals at δ 6.73, 7.20, 8.08 and 8.23 ppm, and triplet peak at δ 7.69 ppm were assigned for aromatic protons respectively. The singlet signal at δ 9.37 was assigned to the hydroxyl protons.
The reaction between quercetin dihydrate and p-chlorobenzoic acid in the presence of phosphorous oxychloride and zinc chloride yielded the formation of multiple products. The compound was separated and purified by column chromatography using suitable solvents. Quercetin-3,5-O-4-chlorobenzoic acid ester derivative (QB3) was a pale yellow colour solid (Table 1). The 1H NMR spectra of QB3 showed ten proton peaks. Spectra revealed singlet signals at δ 6.07, 6.25 and 6.54 ppm and doublet signals at δ 6.85, 7.25, 7.51, 7.74, 8.18 and 8.35 ppm were assigned to aromatic protons respectively. The singlet peak at δ 9.67 ppm was assigned to the hydroxyl protons. As per Scheme 1, quercetin dihydrate was treated with p-methylbenzoic acid in the presence of phosphorous oxychloride catalysed by zinc chloride to form multiple products that was separated and purified by column chromatography to afford compound quercetin-3-O-4-methylbenzoic acid ester derivative (QB4) as a pale-yellow solid. The 1H NMR spectra of QB4 integrated for ten proton peaks. Spectra displayed characteristic singlet signal at δ 2.61 ppm assigned to CH3 protons. The signals for aromatic protons appeared at δ 6.34, 6.62 and 7.69 ppm as singlet and 7.82, 8.19, 8.40 and 8.73 ppm as doublet. The singlet signals at δ 10.16 and 12.18 were assigned to the hydroxyl protons.
Physicochemical and Drug-likeness Properties
An attempt was made to determine the chance for synthesized quercetin derivatives to become an oral drug with the bioavailability of the molecule by analysing physicochemical property, water solubility, lipophilicity, pharmacokinetic and drug-likeness properties. These physicochemical parameters are for predicting the aqueous solubility, intestinal permeability and oral bioavailability of synthesized compounds.
In pharmacokinetic property screening and prediction, the synthesized derivatives should satisfy the binding affinity and ADMET screening selection criteria with eligible water solubility, high GI absorption, eligible drug-likeness and low hERG inhibition risk.17 All the other chemical compounds were excluded due to unsuitable water solubility, GI absorption, drug-likeness and toxicity. The predicted results are summarized in Table 2.
The quercetin derivatives QB2 and QB3 were excluded due to high hERG inhibition risk, low GI absorption and violations of Lipinski’s rules. The molecule QB and QB1 was included due to high GI absorption, no violations of Lipinski’s rules and medium hERG inhibition risk whereas as the QB4 was included for the reasons of GI absorption, one violation of Lipinski’s rules ie., Egan violations, and medium hERG inhibition risk. The detailed information such as pharmacokinetic property screening and prediction of synthesized quercetin derivatives are summarized in Table 2.
Biological activities
In vitro antioxidant activity
In vitro antioxidant activity of synthesised compounds was performed on H2O2 and DPPH free radicals. Ascorbic acid was used as a standard. The IC50 values in H2O2 and DPPH assays for the tested compounds were shown in Table 3. The IC50 values of the compounds suggested that the compound QB1 and QB2 have shown potent activity for both H2O2 and DPPH scavenging and better activity than the standards used.
In vitro cytotoxicity studies
The synthesised compounds were evaluated for in vitro cytotoxicity against MCF-7 breast cancer cell lines using MTT assay. The percentage of cell viability was calculated and concentration of test samples of synthesised compounds needed to inhibit cell growth by 50% values were calculated from the dose-response curves. The CTC50 values for the tested compounds have been shown in Table 4. The CTC50 values showed that among all the synthesised compounds, QB2 showed remarkable cytotoxicity on breast cancer cells of MCF-7 when compared to 5-FU. In general, all the four compounds have shown potent activity against MCF-7 cells.
Molecular Docking studies
The four designed ligands and quercetin were docked with 4NOS protein. Docking was performed using CDOCKER algorithm in which docking estimation is done by calculating the – CDOCKER energy (based on receptor – ligand interaction energy and internal ligand strain energy) and – CDOCKER interaction energy (non-bonding interaction energy existing between the protein and ligand). Greater value of –CDOCKER energy and –CDOCKER interaction energy is more favourable binding interaction between ligand and the protein.
All the ligands efficiently docked with the protein target and displayed good – CDOCKER interaction energies (12.97 to 64.69 kcal/mol) as shown in Table 3. As shown in Fig. 2A and 3A, the catechol hydroxyl group at 4' position of quercetin formed hydrogen bond with ARG B:381 with bond length of 5.54 Å. Pi-alkyl interactions with ILE B-462, MET B:120 and VAL B-465 residues were seen in quercetin. The –CDOCKER interaction energy was 23.31 kcal/mol and –CDOCKER energy was 20.56 kcal/mole for quercetin.
In molecular docking interaction study, QB1 showed –CDOCKER interaction energy value of 57.99 kcal/mol and –CDOCKER energy value of 48.72 kcal/mol. Conventional hydrogen bond interaction with TYR A:491, PRO A:350 and TRP A:372, Pi-Pi stacked and Pi-Pi T-shaped interaction with PHE A:369 and TRP A:194 and Pi-alkyl interaction with PRO B:467 and MET B:120, as shown in figure 2B and figure 3B (Table 5). Among all the ligands, QB2 showed highest value of –CDOCKER interaction energy value of 64.69 kcal/mol and –CDOCKER energy value of 37.03 kcal/mol. In figure 2C and 3C, the conventional hydrogen bond formation with TYR A:491, Pi-Pi stacking interaction with TRP A:463, PHE A:369 and TRP A-194, Pi-alkyl interaction with ALA A:197, PRO A:350 and MET A:355, and van der Waals interaction with PHE A:488, TYR A:489, LEU A:209, TRP A:372, TYR A:373, GLY A:202 was found and has shown better interaction energy when compared with other quercetin derivatives. QB3 displayed –CDOCKER interaction energy value of 60.29 kcal/mol and –CDOCKER energy value of 48.28 kcal/mol. From Figures 2D and 3D, it has seen that the carbonyl oxygen of ester moiety formed conventional hydrogen bond with ILE A:201 residue. Pi-sulfur interaction was found with TRP A:372 and PHE A:369 residue, Pi-alkyl interaction with ARG A:199, LEU A:125, TYR A:491, PHE A:488, MET A:374, PRO A:467, ARG A:381, and ALA A:197 residues.
Further, the –CDOCKER interaction energy and –CDOCKER energy for QB4 displayed was 51.33 kcal/mol and 40.32 kcal/mol respectively. Conventional hydrogen bond is formed with TYR A:373 and TYR A:491, Pi-anion interaction with GLU A:377 and ARG A:199, Pi-alkyl interactions with VAL A:352 and TRP A:372, and van der waals interaction with ASP A:382, TRP A:346, GLN A:263, ALA A:351, TRP A:463, MET A:355 (Figure 2E and 3E). Lastly, the docking study of standard drug 5-FU was observed and found to be conventional hydrogen bond with ARG A:381, TRP A:463 and Pi-sulfur bond with MET A:120.
Novel quercetin analogues were synthesised from quercetin by one pot synthesis, purified and characterised. Physicochemical and drug-likeness properties were concluded for the eligibility of oral drug. The in vitro antioxidant and cytotoxicity studies, IC50 values revealed that compound QB1 was potent antioxidant in both H2O2 scavenging and DPPH assay methods and the results were greater than those of the standards used. Compound QB2 highest cytotoxicity against MCF-7 cancer cell line and also best docked ligand with good interaction energy and favourable interaction with the target enzyme 4NOS. We conclude from the results of biological activities and docking studies that the QB1 and QB2 compounds can be further analysed and optimised as the future potential drug candidates for cancer treatment.
Acknowledgements
Authors sincerely thank JSS College of Pharmacy, Mysuru, Karnataka for providing the laboratory facility to carry out the experimental work. Department of Biotechnology, JSS College of Pharmacy, Ooty is greatly acknowledged for conducting in vitro cytotoxicity studies.
- Sharma N (2014) Free radicals, antioxidants and disease. Biol Med 6:1–6. https://doi.org/10.4172/0974-8369.1000214
- David AVA, Arulmoli R, Parasuraman S (2019) Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacogn Rev 10:84. https://doi.org/10.4103/0973-7847.194044
- Panche AN, Diwan AD, Chandra SR. Flavonoids: An overview, J Nutr Sci 5 (2016). https://doi.org/10.1017/jns.2016.41
- Chen X, Yin OQ, Zuo Z, Chow MS (2005) Pharmacokinetics and modelling of quercetin and metabolites. Pharm Res 22:892. https://doi.org/10.1007/s11095-005-4584-1
- Rossi M, Rickles LF, Halpin WA (1986) The crystal and molecular structure of quercetin: A biologically active and naturally occurring flavonoid. Bioorg Chem 14:55. https://doi.org/10.1016/0045-2068(86)90018-0
- Cárdenas M, Marder M, Blank VC, Roguin LP (2006) Antitumor activity of some natural flavonoids and synthetic derivatives on various human and murine cancer cell lines. Bioorg Med Chem 14:2966. https://doi.org/10.1016/j.bmc.2005.12.021
- Danihelová M, Veverka M, Šturdík E, Jantová S (2013) Antioxidant action and cytotoxicity on HeLa and NIH-3T3 cells of new quercetin derivatives. Interdiscip Toxicol 6:209. https://doi.org/10.2478/intox-2013-0031
- Lu C, Huang F, Li Z, Ma J, Li H, Fang L (2014) Synthesis and Bioactivity of Quercetin Aspirinates. B Korean Chem Soc 35:518. https://doi.org/10.5012/bkcs.2014.35.2.518
- Zhong D, Liu M, Cao Y, Zhu Y, Bian S, Zhou J, Wu F, Ryu KC, Zhou L, Ye D, Discovery of metal ions chelator quercetin derivatives with potent anti-HCV activities, Molecules 20 (2015) 6978. https://doi.org/10.3390/molecules 20046978
- Gurunadham G, Raju RM, Venkateswarlu Y (2015) Simple and efficient esterification reaction catalyzed by Zinc chloride (ZnCl2). Int J Sci Res Publ 5:1
- Khan MF, Nahar N, Rashid RB, Chowdhury A, Rashid MA (2018) Computational investigations of physicochemical, pharmacokinetic, toxicological properties and molecular docking of betulinic acid, a constituent of Corypha taliera (Roxb.) with Phospholipase A2 (PLA2). BMC Complement Altern Med 18(1):1
- Badami S, Prakash O, Dongre SH, Suresh B (2005) In vitro antioxidant properties of Solanum pseudocapsicum leaf extracts. Ind J Pharmacol 37:251. https://doi.org/10.4103/0253-7613.16573
- Jayaprakasha GK, Rao LJ, Sakariah KK (2004) Antioxidant activities of flavidin in different in vitro model systems. Bioorg Med Chem 12:5141. https://doi.org/10.1016/j.bmc.2004.07.028
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 48(3):589
- Hoang TK, Huynh TK, Nguyen TD (2015) Synthesis, characterization, anti-inflammatory and anti-proliferative activity against MCF-7 cells of O-alkyl and O-acyl flavonoid derivatives. Bioorg Chem 63:45. https://doi.org/10.1016/j.bioorg.2015. 09.005
- Singh SP, Konwar BK (2012) Molecular docking studies of quercetin and its analogues against human inducible nitric oxide synthase. Springerplus 1:1. https://doi.org/10.1186/2193-1801-1-69
- Sun Y, Yang AW, Hung A, Lenon GB, Screening for a Potential Therapeutic Agent from the Herbal Formula in the 4th Edition of the Chinese National Guidelines for the Initial-Stage Management of COVID-19 via Molecular Docking. Evidence-Based Complementary and Alternative Medicine (2020) (2020) 1. https://doi.org/10.1155/2020/3219840
Due to technical limitations, table 1 to 5 is only available as a download in the Supplemental Files section.
- Tables.docx
- Scheme01.png
Synthesis of quercetin ester derivatives using aromatic acids.
{kind=link}