The effect of high temperature on bud outgrowth in ‘SEI NO ISSEI’
Decapitated ‘SEI NO ISSEI’ plants exposed to the 28°C/23°C treatment presented an acrotonic pattern of bud out-growth along their stems, whereas those exposed to 38°C/33°C produced very few buds (Fig. 1a). After a four day exposure to two temperature regimes (28°C/23°C or 38°C/33°C), there was no difference in the bud burst rate. However, when decapitated chrysanthemum was exposured to 28°C/23°C or 38°C/33°C on the seventh day, the bud burst rate was significantly different in the fifth and sixth nodes (the first node n-1 refers to the first bud of decapitated chrysanthemum from top to bottom). Buds formed by plants exposed for ten days to 28°C/23°C exhibited a significantly higher bud burst rate than those of plants exposed to 38°C/33°C (Fig. 1b). In the former plants, bud growth was acrotonic, as also shown by the pattern of leaf development, whereas bud growth under the higher temperature regime was inhibited and no acrotonic gradient was observed. The length of a bud of different position was two to twelve fold greater in plants exposed for ten days to the lower temperature regime than in those exposed to the higher temperature regime (Fig. 1c).
Transcriptome sequencing and read assembly
In order to study the differentially expressed genes of buds of decapitated ‘SEI NO ISSEI’ between 38°C/33°C and 28°C/23°C, four cDNA libraries were generated using the RNA-Seq platform, i.e., h-24 h (38/33°C for 24 h), h-96 h (38/33°C for 96 h), n-24 h (28/23°C for 24 h) and n-96 h (28/23°C for 96 h). After filtering the raw reads, we obtained 44.13 Mb, 44.52 Mb, 44.63 Mb and 45.46 Mb clean reads from four cDNA libraries, containing 6.62 Gb, 6.68 Gb, 6.69 Gb and 6.82 Gb clean bases, respectively. The clean reads ratios, the Q20 percentages, the Q30 percentages of the four samples were more than 83%, 98% and 94%, respectively (Table 1). Using Tgicl [26] to cluster the transcripts, we obtained 132,396 unigenes with the total length of 140,318,943 bp, the mean length of 1,059 bp and the GC percentage of 39.48% (Table 2).
Unigene functional annotation
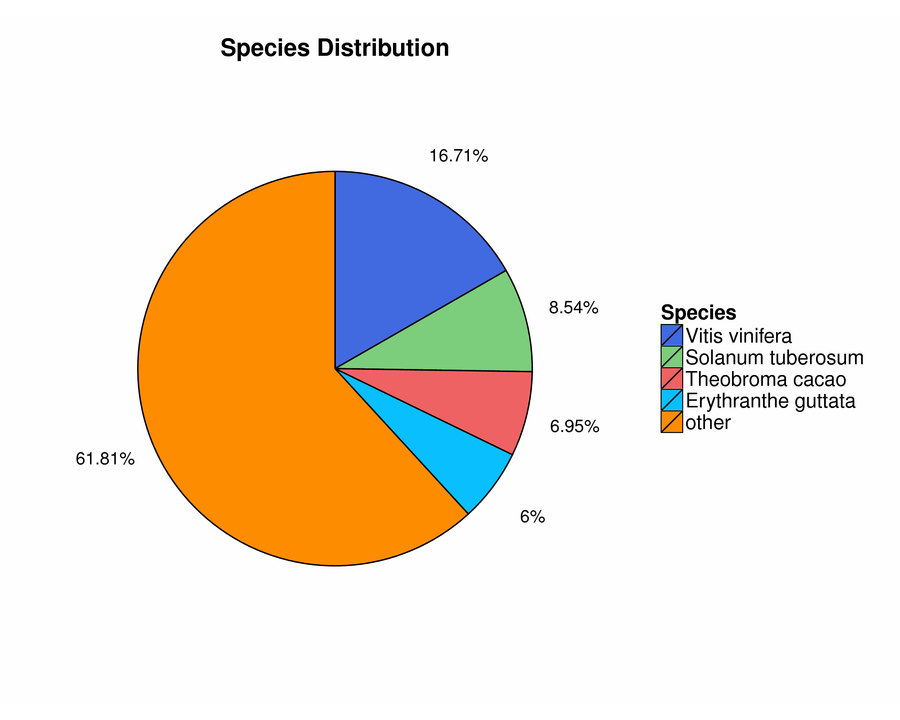
To predict the potential functions of unigenes in the ‘SEI NO ISSEI’ buds, after assembly, we performed seven functional database annotations (NR, NT, Swiss-Prot, KEGG, COG, Interpro and GO). 132,396 unigenes were assembled, of which 79,116 unigenes were annotated in the seven functional databases. The number of unigenes annotated in the NR, NT, Swiss-Prot, KEGG, COG, Interpro, and GO databases were 72,274, 56,878, 51,697, 55,399, 27,433, 49,316 and 17,244, respectively, meanwhile, the percentages of annotations were 54.59%, 42.96%, 39.05%, 41.84%, 20.72%, 37.25%, and 13.01%, respectively (Table 3). Among them, the NR database had the largest number of unigenes, so we counted the species distribution according to the NR annotation results. The most common species was Vitis vinifera, which accounted for 16.71% of species distribution, followed by Solanum tuberosum (8.54%), Theobroma cacao (6.95%) and Erythranthe guttata (6.00%). The proportion of other species was 61.81% (Figure S1).
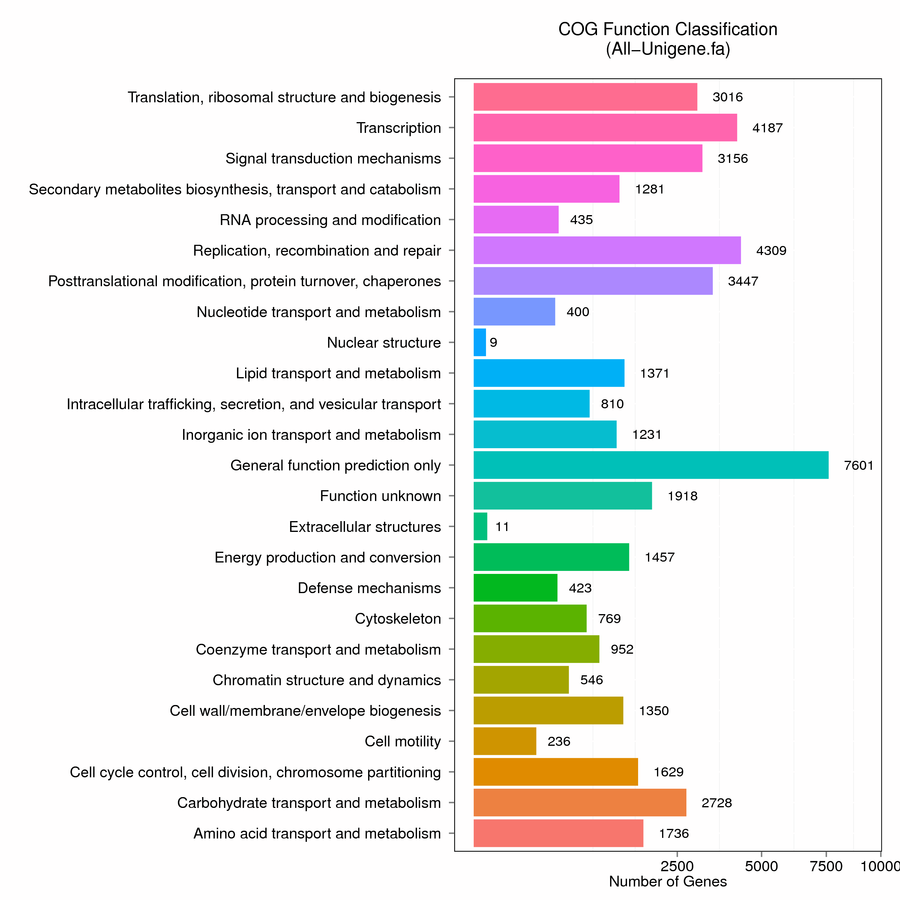
To clarify the potential functions of unigenes in the buds of ‘SEI NO ISSEI’, we calculated its functional classification based on the COG annotation results. The COG annotation divided unigenes into 25 categories, the most of which was “general functional prediction only” (7601, 16.89%), followed by “replication, recombination and repair” (4309, 9.57%) and “transcription" (4187, 9.30%). However, the least two categories were "nuclear structure" and "extracellular structures", accounting for 0.019% and 0.024% of unigenes, respectively (Figure S2).
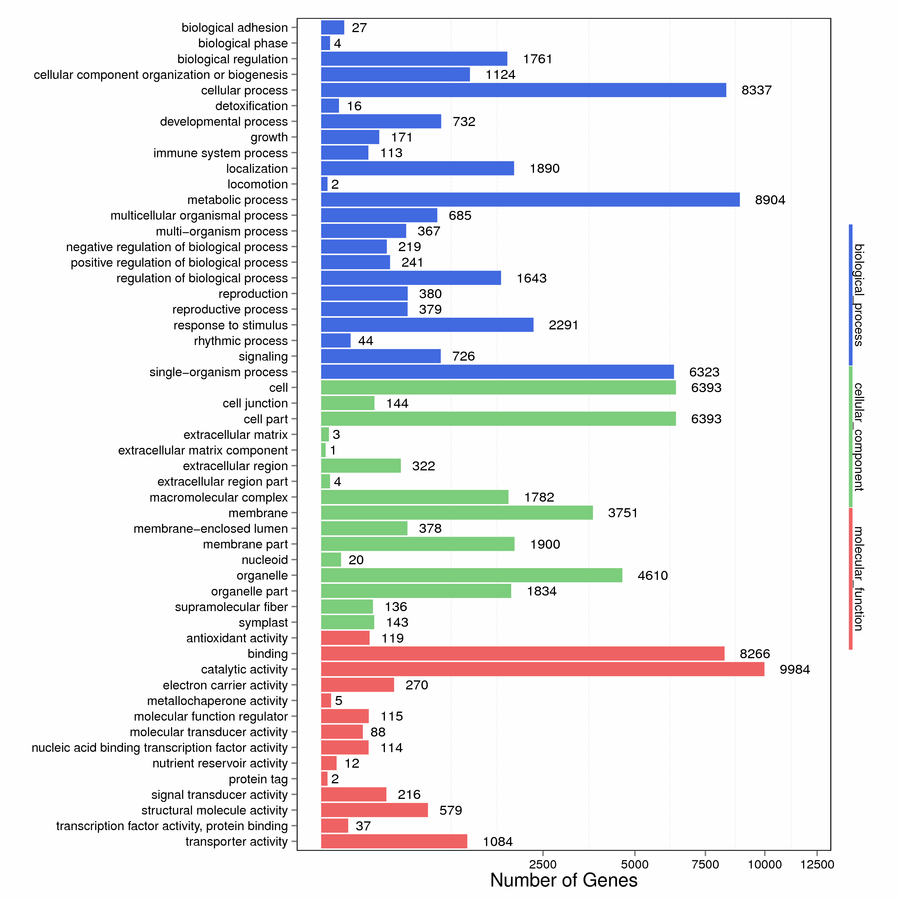
To further understand the potential functions, GO term enrichment analysis was performed. A total of 85,084 unigenes were divided into 53 functional groups, which mainly included biological process (23), cellular component (16) and molecular function (14). In biological process, the main functional groups were “metabolic process” (8904) and “cellular process” (8337). In cellular component, the main functional groups were “cell” (6393) and “cell part” (6393). Among the molecular function, "catalytic activity" (9984) and "binding" (8266) were most (Figure S3). The results suggested that these main categories might play an important role in buds.
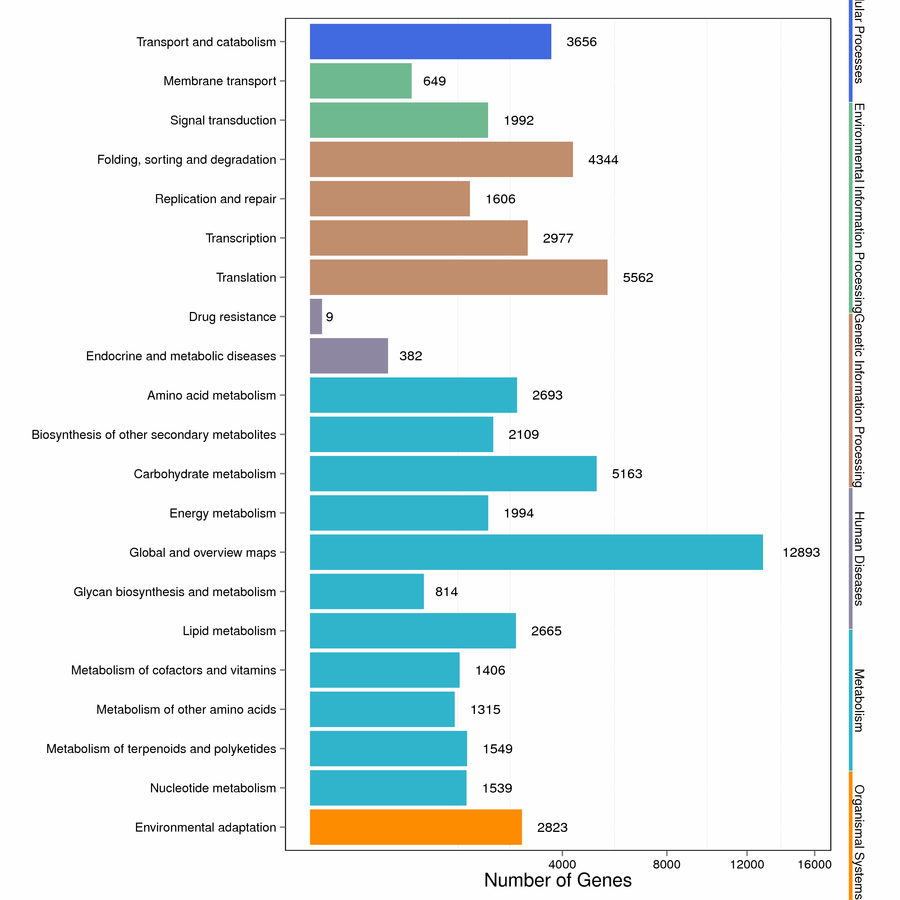
In addition, we calculated its functional classification based on the results of the KEGG database. A total of 58,140 unigenes were clustered, including cellular processes, environmental information processing, genetic information processing, human diseases, metabolism and organismal systems. The three main pathways were the “global and overview maps” in metabolism (12,893, 22.18%), the "translation" in genetic information processing (5,562, 9.57%), and the "carbohydrate metabolism" (5,163, 8.88%) (Figure S4).
Identification of transcription factors (TFs) involved in buds under contrasting temperature regimes
Multiple differentially expressed TFs were identified in buds of decapitated ‘SEI NO ISSEI’ under 28°C/23°C or 38°C/33°C. Most transcription factors belonged to the AP2/ERF, MYB, WRKY and bHLH families. We analyzed 8 AP2/ERFs, 6 MYBs, 3 WRKYs and 1 bHLH (Fig. 2 and Table S2). In the AP2/ERFs family, the expression levels of CL5614.Contig4_All (RAP2-7), CL2842.Contig2_All (ERF12), Unigene38388_All (ERF13), Unigene38278_All (ERF53) and Unigene28455_All (ERF110) were significantly higher in h-96h than n-96h, however, the expression levels of Unigene34736_All (WIN1), CL5120.Contig2_All (ERF017) and Unigene69087_All (RAP2-7) were significantly inhibited at 96 h after high temperature treatment (Fig. 2 and Table S2). In the MYBs family, Unigene 5596_All (MYB46), Unigene9775_All (MYB4), CL10164.Contig2_All (MYB6), CL262.Contig9_All (MYB) and CL1495.Contig1_All (MYB44) were induced more strongly in h-96h than in n-96h. CL6283.Contig2_All (MYB6) was more suppressed in 96 h after high temperature treatments (Fig. 2 and Table S2). In the WRKYs family, CL3506.Contig5_All (WRKY33) and CL2694.Contig5_All (WRKY40) were all more up-regulated during 38°C/33°C and down-regulated during 28°C/23°C, however, CL3506.Contig11_All (WRKY33) was inhibited in h-96h (Fig. 2 and Table S2). In the bHLH family, bHLH-related gene (CL3647.Contig6_All) was more up-regulated in 38°C/33°C than in 28°C/23°C (Fig. 2 and Table S2). DEGs suggested that these TFs played important roles in the buds of the decapitated 'SEI NO ISSEI' under contrasting temperature conditions.
Differentially transcribed of auxin-related genes in buds
Auxin plays a key role in bud growth and development, we analyzed auxin-related differential genes from RNA-seq data. There were 32 differential transcriptions associated with auxin, 21 genes in the signalling pathway and 11 genes in the transport pathway (Fig. 3 and Table S3). Among 21 transcripts associated with auxin signalling process, the abundance of TIR1, ARF2, ARF4, ARF5, ARF8, ARF16, ARF18, ARF19, IAA3 and IAA9 transcript were low in buds outgrowth (n-24h and n-96h) compared to other time points (h-24h and h-96h), while PIN1, PIN2, AUX1, LAX1, LAX2 and ABCB1 (associated with auxin transport process) were significantly up-regulated in buds outgrowth compared to inhibited buds.
Verification of RNA-seq data by qRT-PCR
In the library of bud outgrowth, fourteen differentially expressed genes were selected for qRT-PCR to test the reliability of RNA-seq data. The qRT-PCR assays largely validated the RNA-Seq based identification of differential transcription (Fig. 4 and 5). We selected four transcription factors, including Unigene28455_All (ERF110), Unigene9775_All (MYB4), CL2694.Contig5_All (WRYY40) and CL3647.Contig6_All (bHLH36) and ten auxin-related genes (TIR1, ARF2, ARF16, IAA3 and IAA9), which were all related to shoot branching and potential candidate genes for regulating chrysanthemum branching.
The phenotype of A. thaliana plants heterologously expressing CmERF110
When CmERF110 was constitutively expressed in A. thaliana, the number of branches formed by 45 day old plants varied between wild type (WT) plants, WT plants harboring the transgene p35S::CmERF110 (WT/ERF110), brc1 mutant plants and brc1 mutant plants harboring p35S::CmERF110 (brc1/ERF110): the latter genotype produced the highest number of branches, followed by WT/ERF110 and brc1 (Fig. 6a and 6b). Axillary buds were rarely formed in the first three rosette leaf axils of WT plants, whereas nearly all rosette leaves carried buds or branches in the axils of WT/ERF110 (active buds), brc1 (inactive buds) and brc1/ERF110 (active buds) plants. A greater number of high order branches was produced by WT/ERF110, brc1 and brc1/ERF110 plants than by WT plants (Fig. 6g-j). WT/ERF110, brc1 and brc1/ERF110 plants formed a similar number of primary cauline leaf branches (CI) as did WT ones (Fig. 6k). WT/ERF110 and brc1/ERF110 plants produced significantly more secondary cauline leaf branches (CII) than did, respectively, WT and brc1 plants (Fig. 6l). The brc1/ERF110 plants formed more rosette branches (RI and RII) than did WT plants. The RI and RII phenotype of brc1 plants was weaker than that of WT/ERF110 (Fig. 6m and 6n). The conclusion was that the product of BRC1 influenced the plants’ RI and RII performance, while the constitutive expression of CmERF110 mainly affected CII as well as RI and RII.
The constitutive expression of CmERF110 in A. thaliana reprogrammed the transcription of auxin-related genes
The effect of constitutively expressing CmERF110 on the transcription of the auxin-related genes was tested in 21 day old A. thaliana seedlings. The abundance of PIN1, AUX1, LAX1, LAX2 and ABCB1 transcript was from 1.30 to 3.14 fold higher in the transgenic plants than in WT plants, while TIR1, ARF2, ARF16, IAA3 and IAA9 were down-regulated by between 1.23 and 1.33 fold (Fig. 7). The suggestions that in the transgenic plants, the CmERF110 product participated in the shoot branching, acting through the auxin-related genes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}