Single-crystal X-ray diffraction studies revealed that both Sn2(dobdc) and Sn2(dobpdc) crystallize in the monoclinic P21/c space group (Supplementary Table 2). The framework of Sn2(dobpdc) is analogous to Sn2(dobdc), featuring the same coordination spheres of Sn2+ ions but an enlarged spacing between the Sn2+ ions due to the extended dobpdc4− linker. The structure of Sn2(dobpdc) is described here representatively. There is one crystallographically independent Sn2+ ion that lies in a nonsymmetric disphenoidal coordination environment and is coordinated by four oxygen atoms from three different dobpdc4− ligands (Supplementary Fig. 1a). The Sn–O bond lengths are in the range from 2.065(6) to 2.181(3) Å. The selected bond lengths and angles are provided in Supplementary Table 3, which are in the range of other types of Sn-based coordination compounds.33 The Sn2+ ions are bridged by µ2-η1:η1 carboxylate groups into Sn–COO chains along the b axis, which are further connected via dobpdc4− to form a 3D framework (Supplementary Figs. 1b and 1c). Topologically, the organic ligand and Sn2+ ion can be viewed as six- and three-connected nodes, respectively. Thus, the framework can be viewed as a (3,6)-connected rtl net (Supplementary Fig. 2).34 Due to the expanded linkers, the distances between the Sn–COO chains increase from 8.8672(12) Å for Sn2(dobdc) to 13.1427(16) Å for Sn2(dobpdc).
Both Sn2(dobdc) and Sn2(dobpdc) maintain their phase purity and structural stability after immersion in the electrolyte for 7 days, demonstrating high electrolyte resistance (Supplementary Fig. 3). Thermogravimetric analysis showed that Sn2(dobdc) and Sn2(dobpdc) are stable up to 500 °C under a nitrogen atmosphere (Supplementary Fig. 4). The thermal stabilities of Sn2(dobdc) and Sn2(dobpdc) were further evaluated by in situ variable-temperature PXRD measurements on a Pt sample platform in the temperature range from 50 to 600 °C (Supplementary Fig. 5). The PXRD patterns for both MOFs remain unchanged until 500 °C, further demonstrating their excellent thermal stability. The high stabilities of Sn2(dobdc) and Sn2(dobpdc) are uncommon for Sn-based coordination compounds due to the strong coordination bonds and 3D rigid framework structures, which are suitable for lithium storage studies.
Taking advantage of the multiple lithium storage active sites from the alloying-type Sn centers and electrochemically active groups of organic ligands, Sn2(dobdc) and Sn2(dobpdc) were evaluated as anode materials for LIBs in CR2032 coin-type cells at 298 K. According to the reversible reaction of Sn-based oxides with lithium10, the electrochemical process of Sn-MOFs with lithium is expected to undergo a combination of electrochemical conversion and alloying mechanisms, with the following two steps: Sn2L + 4Li+ + 4e− ↔ 2Sn + Li4L (L4− = dobdc4− or dobpdc4−) and Sn + xLi+ + xe− ↔ LixSn (0 ≤ x ≤ 4.4). To investigate this redox chemistry for lithium storage, the cyclic voltammetry (CV) curves for both MOFs were measured at a scan rate of 0.1 mV s− 1 between 0.01 and 3.0 V (Figs. 2a and 2b). It is noted that the first cathodic scan with two irreversible cathodic peaks in the ranges of 0.7–1.1 V and 0.3–0.6 V differs from the subsequent scans. This behavior is attributed to the formation of a SEI layer that causes the initial capacity loss.35,36 The activation of Sn-MOFs to form amorphized materials also occurs during the first lithiation process (a detailed analysis is in the in situ PXRD section). After the first cycle, two pairs of distinctive redox peaks are observed, which are attributed to the gradual insertion or extraction of Li+ within the organic ligands and metal centers, respectively. The peak at 0.8 V is due to lithium insertion into the conjugated aromatic ligand with a reduction in the Sn centers, while the peak at 0.2 V is ascribed to the alloying reaction of lithium with Sn. All peaks remain invariable after the first cycle, confirming the presence of reversible and stable electrochemical reactions. Figures 2c and 2d display the first five galvanostatic discharge–charge profiles of the cells at 100 mA g− 1. Sn2(dobpdc) shows a lower discharge plateau than Sn2(dobdc). After the formation of the SEI layers with an initial irreversible capacity loss, both Sn2(dobdc) and Sn2(dobpdc) exhibit stable lithium storage behaviors, indicating few side reactions of the electrolyte. Two sloping discharge plateaus for Sn2(dobdc) are observed in the ranges of 1.2–0.6 V and 0.45 − 0.15 V, and the discharge plateaus for Sn2(dobpdc) are in the ranges of 0.8 − 0.5 V and 0.4 − 0.1 V (Figs. 2c and 2d), which agrees with the electrochemical reactions in the CV results. Figure 2e shows the rate performances after cycling at various current densities from 200 to 2000 mA g− 1. Even at 2000 mA g− 1, the capacities of the electrodes are still much higher than the theoretical capacity of commercial graphite. Figure 2f shows the cycling performance at 200 mA g− 1. After 200 cycles, the Sn2(dobpdc) anode achieves a capacity of 1018 mAh g− 1, whereas Sn2(dobdc) maintains a capacity of 731 mAh g− 1 at 200 mA g− 1. The cycling performances at high currents are also outstanding. At 500 mA g− 1 for 600 cycles, the Sn2(dobdc) and Sn2(dobpdc) electrodes exhibit stable capacities over 400 and 650 mAh g− 1, respectively (Supplementary Fig. 6).
Due to the isoreticular structures of Sn2(dobdc) and Sn2(dobpdc), the distinct lithium storage capacities between Sn2(dobdc) and Sn2(dobpdc) could be derived from the expansion of the organic linkers. Apart from the alloying reactions of the Sn centers, which are almost the same in these two MOFs, density functional theory (DFT) calculations were performed to evaluate the lithium storage capability of organic ligands. Based on the calculated Li uptake numbers of 8 and 12 for the organic ligands (Supplementary Fig. 7 and Supplementary Tables 4 and 5), Sn2(dobdc) and Sn2(dobpdc) are able to deliver theoretical capacities of 1044 mAh g− 1 and 1099 mAh g− 1, respectively, with contributions from the Sn centers. The practical capacities of the Sn2(dobdc) and Sn2(dobpdc) are 731 and 1018 mAh g− 1 at 200 mA g− 1, corresponding to utilization efficiencies of 70.0% and 92.6%, respectively. The high utilization efficiency of Sn2(dobpdc) is attributed to the increased conjugation of the organic ligand, which can realize the full electrochemical activity of both redox active groups and aromatic rings.
To reveal the difference in the rate performances of Sn2(dobdc) and Sn2(dobpdc), which mainly depend on the kinetics during the lithium storage processes, electrochemical impedance spectra (EIS) and additional CV characterization were performed. The activation energies of these two Sn-based MOFs for lithium storage were estimated by the EIS data at different temperatures (Figs. 3a-3c). In general, a semicircle at high and medium frequencies and a straight line at low frequencies are related to the charge transfer resistance (Rct) and the Li+ diffusion resistance (Rw), respectively. The value of Rct decreases as the temperature increases because electrochemical kinetic reactions can easily occur at high temperatures.37 Sn2(dobpdc) exhibits a smaller Rct and a lower Rw than Sn2(dobdc), indicating that Sn2(dobpdc) undergoes faster reaction kinetics (Figs. 3a and 3b). Based on the EIS data at different temperatures, the apparent activation energy (Ea) can be calculated by i0 = RT/nFRct and i0 = Aexp(-Ea/RT),37,38 where i0 is the exchange current, A is the temperature-independent coefficient, R is the gas constant, T is the absolute temperature, n is the number of transferred electrons, and F is the Faraday constant. The Ea value is obtained by the slope of the ln(T/Rct) versus 1000/T plot fitted by the Arrhenius equation (Fig. 3c). The calculated Ea values are 62.68 kJ mol− 1 and 53.46 kJ mol− 1 for Sn2(dobdc) and Sn2(dobpdc), respectively. The relatively low Ea of Sn2(dobpdc) indicates an easier diffusion process for lithium intercalation than that in Sn2(dobdc). Figure 3d displays the EIS of these two Sn-MOFs after 200 cycles. The Sn2(dobpdc) electrode exhibited a lower Rct of 71.8 Ω in comparison to the Rct of 175.4 Ω for the Sn2(dobdc) electrode, in accordance with the faster reaction kinetics of Sn2(dobpdc). Furthermore, the CV tests at different scan rates (0.1–0.4 mV s− 1) with a voltage range from 0.01 to 3.0 V were also carried out to probe the kinetic origin of the lithium storage in the Sn2(dobdc) and Sn2(dobpdc) electrodes (Supplementary Figs. 8a and 8b). The relationship between the log (peak current) and log (scan rate) from the CV curves suggests that a combination of diffusion-controlled and surface-controlled processes contribute to the electrochemical reactions in the Sn2(dobdc) and Sn2(dobpdc) electrodes (Supplementary Figs. 8c and 8d).10 Quantitative analyses demonstrate that the percentage of capacitive contribution of the Sn2(dobpdc) electrode is higher than that of the Sn2(dobdc) electrode (Supplementary Figs. 8e and 8f), confirming that the Sn2(dobpdc) electrode has favorable charge transfer kinetics and good rate capability. Furthermore, the diffusion coefficients of Li+ in Sn2(dobdc) and Sn2(dobpdc) were calculated by the galvanostatic intermittent titration technique (GITT) (Supplementary Fig. 9), demonstrating that Sn2(dobpdc) has a higher Li+ diffusion coefficient than Sn2(dobdc). The enhanced kinetics of Sn2(dobpdc) are from the high surface area and pore volume of the expanded framework (Supplementary Fig. 10). Therefore, the expanded organic ligand can not only enhance π-aromatic conjugation but also improve the contact area between the electrode and electrolyte, resulting in a high utilization of active sites for a high capacity (Fig. 2f) and fast kinetics for lithium storage (Fig. 2e).
To determine the lithium storage mechanism of these Sn-based MOFs, in situ and ex situ characterizations were performed to analyze these Sn-MOF-based electrodes during lithiation and delithiation processes (Figs. 4–6, Supplementary Figs. 11–18). Supplementary Fig. 11a and Fig. 4a are the discharge–charge curves for Sn2(dobdc) and Sn2(dobpdc) at 50 mA g− 1 during the first cycle, respectively, corresponding to the in situ states for the PXRD measurements, as shown in Supplementary Fig. 11b and Fig. 4b, indicating that the crystallinities of Sn2(dobdc) and Sn2(dobpdc) decrease during the first lithiation process. No obvious PXRD peaks are observed after the second lithiation and delithiation processes, revealing that both of them lose the long-range ordered structure and become amorphous upon cycling (Supplementary Fig. 12). Ex situ FTIR spectra were measured to further study the formation of the amorphous MOFs during the first cycle (Figs. 4c-4e and Supplementary Figs. 11c-11e). The selected states of the Sn2(dobpdc) electrode at 100 mA g− 1 are marked in Fig. 4c. For the fresh electrode at open-circuit voltage (Fig. 4d), the infrared (IR) peaks at approximately 1640, 1390, and 1080 cm− 1 are assigned to the asymmetric stretching vibration of COO−, the symmetric stretching vibration of COO− and the stretching of C–O bonds, respectively, under the coordination effects of Sn ions.39,40 When the potential drops to 1.0 V for a specific capacity of approximately 90 mAh g− 1, the corresponding FTIR spectra at the a and b states are congruent with the initial states, in accord with the crystalline stability after initial discharge to 1.0 V from the in situ PXRD results. After being fully discharged to 0.01 V, the characteristic absorbance peaks of the COO− and C–O groups become relatively weak, indicating that the COO− and C–O groups are the corresponding active sites for lithiation. After being fully charged to 3.0 V, three characteristic absorbance peaks at 1640, 1390 and 1080 cm− 1 are recovered, indicating that the coordination units of amorphous Sn2(dobpdc) are maintained after the first cycle.41 The changes in the FTIR peaks for the Sn2(dobdc) electrode are similar to those for the Sn2(dobpdc) electrode (Supplementary Figs. 11c-11e). The amorphization during the first discharge process could endow these active materials with an elastic strain capability to accommodate the volume variation from the alloying reaction of Sn with lithium, which is consistent with the excellent cycling performances of Sn2(dobdc) and Sn2(dobpdc) (Fig. 2f). In addition, due to the locally remaining coordination structure, these amorphized samples with crystal defects demonstrate a redox active performance that is superior to that of their crystalline counterparts (Fig. 4e).
To study the valence state and local coordination structure evolution around Sn centers in the amorphous materials during cycling, quasi-in situ XAFS spectra were measured corresponding to the marked states, as shown in Fig. 5a, by which X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS)42 could provide fundamental information about the valence state and local coordination structure of the Sn2(dobpdc) electrode (Fig. 5 and Supplementary Figs. 13, 14), respectively. The XAFS spectra of the pristine Sn2(dobpdc) electrode confirmed the bivalent state of Sn2+ and Sn–O coordination of 2.13 Å with a coordination number (CN) of 3.7±0.7 (Supplementary Fig. 13 and Table 6), which is consistent with the crystal data of Sn2(dobpdc) (Supplementary Table 3). The quasi-in situ tests were performed on a treated CR2032 coin battery (Fig. 5b). During the discharge process (Fig. 5c), the amplitude of the white line in the XANES profile of the Sn2(dobpdc) electrode slowly decreases as a result of the reduction of Sn centers. During the charge process, the intensity of the main XANES peak of the Sn2(dobpdc) electrode gradually increases back to the original state (Fig. 5d). The insets in Fig. 5c, d are the corresponding Fourier transformation-EXAFS (FT-EXAFS) spectra, which could reveal the local coordination structure change around the Sn centers. Upon discharging, the intensity of the Sn–O bonds continuously decreases due to the insertion of lithium into organic ligands and the alloying reaction of lithium with Sn centers. Moreover, a new peak emerges at approximately 3.0 Å because of the formation of Sn–Sn bonds from the alloying reaction. The Sn–O bonds recover, and the Sn–Sn bonds disappear after fully charging, indicating reversible reactions around the Sn centers. To determine the coordination environment around the Sn center, phase-corrected FT-EXAFS of selected states were fitted in R space. As shown in Fig. 5e and Supplementary Fig. 14, the Sn–O coordination interactions are broken with a decrease in CN of the Sn–O coordination shell from 3.7 ± 0.7 to 2.8 ± 0.5 after being discharged to 1.5 V, corresponding to partial disengagement of the organic ligands from the Sn centers due to lithiation of the organic ligands. Upon becoming fully discharged, a new peak at 2.92 Å is observed and is ascribed to the Sn-Sn shell from the LixSn alloy with a CN of 6.3 ± 1.2, confirming the alloying reaction. During the charging process, the CN of the Sn–Sn shell decreases from 6.3 ± 1.2 to 2.5 ± 0.6 after charging to 0.6 V because of the dealloying reaction of LixSn; after charging to 3.0 V, the Sn–Sn interaction disappears, and the Sn–O shell with a CN of 2.7 ± 0.4 reappears (Fig. 5f) and does not fully return to the original state with a CN of 4 due to the formation of a SEI layer during the initial two cycles. XAFS analyses of the lithiated and delithiated electrodes after 5 cycles were conducted to further reveal the reversibility of the lithium storage mechanism (Supplementary Fig. 14). After five cycles, the XANES curve of the Sn2(dobpdc) electrode in the fully charged state matches well with that of the pristine state, demonstrating the reversible valence state change of the Sn center (Supplementary Fig. 14a). Based on the FT-EXAFS spectra and fitting results (Supplementary Fig. 14b, c and Fig. 5g), a weak Sn–O shell at 2.15 Å with a CN of 1.8 ± 0.3 can be observed in the discharged electrode after 5 cycles, demonstrating the presence of the Sn–O interaction between the LixSn alloy and the organic ligands. In the charged electrode after 5 cycles, the peak at 2.15 Å from the Sn–O shell with a CN of 3.5 ± 0.6 is very close to that of the pristine electrode (3.7 ± 0.7), confirming the reconstruction of the coordination environment of Sn. These results demonstrate the highly reversible transformation between the formation of Sn–O and Sn–Sn bonds during the electrochemical reaction in the Sn2(dobpdc) electrode.
To verify the intermediates that form during the alloying reaction around Sn centers, HRTEM analysis of Sn2(dobpdc) was performed during the second cycle (Fig. 15). Since these MOFs are easily damaged by the electron beam, the lattice fringe information disappears for the pristine states (Supplementary Fig. 16). During the discharge process, the interplanar distances of 0.291, 0.282 and 0.198 nm are consistent with the (200) and (101) lattice planes in Sn and the (933) lattice planes in Li4.4Sn, respectively, confirming the formation of Sn and Li4.4Sn. These results are consistent with the Li+ ions inserted into the active materials with a reduction of Sn(II) to Sn(0) followed by the alloying reaction (Fig. 15a). During the charging process, the appearance of the interplanar distance of 0.294 nm for the (200) lattice planes of Sn metal confirms the dealloying reaction of Li4.4Sn; after becoming fully charged, the amorphous Sn2(dobpdc) recovers with the absence of lattice fringe information (Fig. 15b). These HRTEM results confirm the reversible alloying and dealloying of Sn with Li+ ions.
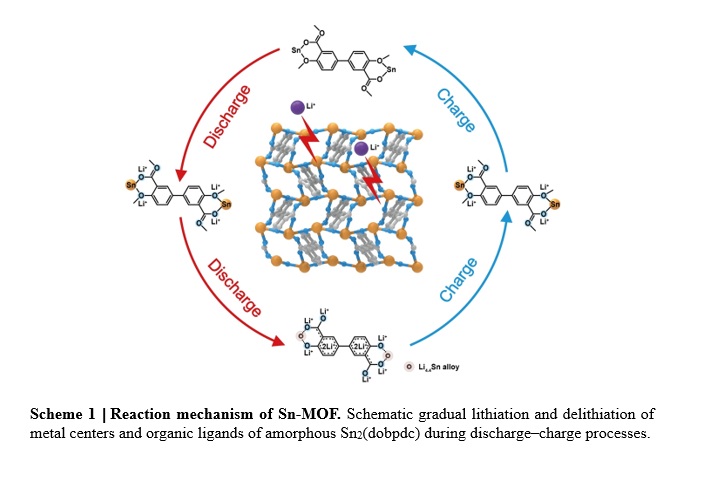
Based on the above analyses, the overall lithiation and delithiation reaction processes of Sn2(dobpdc) can be described, as shown in Scheme 1 (see Supplemental Files for Scheme 1). Although the amorphization of Sn2(dobpdc) occurs during the first cycle, the coordination interaction between Sn centers and organic ligands could be maintained to sustain stable and long-term lithium storage behavior. In the following processes, the electrochemical reactions in the amorphous Sn2(dobpdc) phase are related to two successively reversible processes: the redox reaction of organic ligands and the alloying reaction of Sn centers. During the discharge process, a capacity of ~ 210 mAh g− 1 over the voltage range from 3.0 to 0.5 V is related to Li+ insertion into organic ligands with a reduction of the Sn centers; after becoming fully discharged, the superlithiation of organic ligands and alloying reaction of Sn could provide a capacity of ~ 890 mAh g− 1 for an uptake of approximately 17-Li+. During the charge process, the dealloying of Li4.4Sn to Sn and the extraction of Li+ from organic ligands successively take place, resulting in the reversion of amorphous Sn2(dobpdc) for a reversible specific capacity of ~ 1100 mAh g− 1. Taking advantage of the multiple lithium storage sites and the reversible formation of the coordination bonds, this Sn-based MOF exhibits a high utilization of active sites and fast kinetics for the merits of high reversible capacity at high rate and excellent cycling stability.
We used ex situ FTIR and SEM to study the composition and morphology changes of the electrodes after long-term cycling (Supplementary Figs. 17 and 18). In the FTIR spectra, after the first, fifth and tenth cycles after becoming fully recharged to 3.0 V, the peaks are not entirely consistent with those of the pristine electrodes because of the lithiation and amorphization of the materials, but the FTIR spectra of the fully charged states after five and ten cycles are consistent with that of the first recharged electrode, indicating the recovery of the coordination units. SEM observations show that the morphologies of the active materials are well retained after long-term cycling (Supplementary Fig. 18), accounting for their stable lithium storage performance during cycling.
{kind=link}