MSLN and CLDN6 specific CAR T-cells recognize and kill cognate antigen expressing tumor cell lines.
In this study, we focused on CAR T-cells specific for the tumor antigens mesothelin and claudin 6 (Fig. 1A), which are highly expressed on tumor cells but rarely detected on normal cells. We either used antigen overexpressing cell lines, HeLa (Fig. 1B i) and OVCAR-3 (Fig. 1B ii) for MSLN-specific antigenicity, PA-1 SC12 (Fig. 1B v) and OVCAR-3 (Fig. 1B vi) for CLDN6-reactivity or exogenously overexpressed the antigen on the cell surface of iDCs (MSLN and CLDN6) (Fig. 1B iii & vii) or on antigen-negative cancer cell lines (MDA-MB-231) (Fig. 1B vi) via IVT-RNA electroporation.
We activated CD4+/CD8 + T-cells and transfected them with MSLN (15 µg) and CLDN6 (10 µg) CAR encoding IVT RNAs. At this high dose, we could monitor around 50 % CAR expression for both antigen specificities (Fig. 1C i/D i).
To confirm that the CAR MSLN T-cells are functional in vitro we performed multiple assessments. In the first step, IFN-γ secretion was examined against differenT-cell lines: CAR MSLN T-cells secreted high level of IFN-γ towards MSLN expressing cell lines, up to 4000 pg/mL in case of OVCAR-3 and 1500 pg/mL in case of HeLa cell line as target cell (Fig. 1C ii). There was no significant secretion of this cytokine against the negative control cell lines SK-OV-3 and COLO-699-N. Furthermore, cell cytotoxicity was evaluated by means of a 3D culture spheroid lysis assay (Fig. 1C iii). Solid phase spheroid cells reflect the in vivo situation in some aspects such as actively dividing tumor cells at its boundaries with optimal nutrients provision, and at its core tumor cells becoming apoptotic or necrotic [17]. However, it does not embrace immunosuppressive and angiogenetic effects in TME. Our data confirmed that the CAR MSLN T-cells successfully kill the tumor spheroids even at a low E:T ratio of 1:1, which could achieve almost 100% killing in 24 hours as opposed to mock T-cells. Decrease of fluorescence in negative controls reflects the degradation of GFP encoding mRNA over time. Conclusively, these data proved cytokine secretion and cytotoxic function of CAR MSLN T-cells.
To assess CAR CLDN6 functionality, CAR CLDN6 T-cells were cocultured with IVT-RNA electroporated iDCs expressing CLDN6 heterologously at increasing doses for an E:T ratio of 5:1 to quantify secreted IFN-γ in an ELISA. The amount of detected cytokine ranged between 1000 pg/ml for 2 µg electroporated Cl6 and 100 pg/ml for 0.02 µg electroporated CLDN6 (Fig. 1D ii), and hence, decreased dose-dependently.
For the impedance based cytotoxicity assay, we electroporated the SK-OV-3 3 cell line with 2µg CLDN6 RNA and seeded them on xcelligence plates. After 24 hours, the CAR CLDN6 or the mock T-cells were added at an E:T ratio of 5:1 and the impedance changes were recorded over time. Successful killing of CLDN6+ SK-OV-3 3 cells by CAR CLDN6 effector T-cells (70%) was confirmed, while no relevant cytotoxicity was observed for mock T-cells (Fig. 1D iii).
We also evaluated the proliferation of CAR CLDN6 T-cells applying a CFSE dilution based assay in a dose dependent manner. These CAR T-cells divided strongly up to a frequency of 80%, not only in the presence of a high dose of heterologously expressed CLDN6 in iDCs (2 ug), but also at a dose as low as 0.02 ug CLDN6 (70%; Fig. 1D iv). Notably, no proliferation was detectable in the presence of mock-electroporated iDCs. In conclusion, our data confirmed that both CARs specific for MSLN and CLDN6 elicited superior effector functions against antigen expressing tumor cell lines or iDCs.
Exogenous TGFβ impairs CAR T-cell proliferation in vitro
Due to the fact that TGFβ is one of the key regulator of the immune system in the tumor microenvironment, which vastly suppresses CAR T-cell function [9], we hypothesized that one of the hurdles in solid tumor CAR T-cell therapy arise from high levels of TGFβ in TME. To test this hypothesis, we performed a series of in vitro tests to find out if exogenous TGFβ can exert a suppressive effect on our CAR T-cells specific for 2 different tumor antigens. IVT electroporated iDCs (CLDN6/ MSLN) were cocultured with CAR CLDN6 or CAR MSLN T-cells at increasing doses of TGFβ. A broad range of exogenous TGFβ added (5–20 ng/ml) could significantly suppress CAR T-cell functions in terms of cytokine secretion (Fig. 2A) and proliferation (Fig. 2B) which is consistent with previous reports. Secretion of IFN-γ, the most prevailing immunostimulatory T-cells’ cytokine, is very susceptible to even the lowest dose of TGFβ added, dropping to less than 40 % of the IFN-γ secretion in absence of TGFβ. In line to that, proliferation of both MSLN and CLDN6 specific CAR T-cells was reduced to less than of 50 % of their native proliferation (i.e. absence of TGFβ) in the presence of even very low dose of TGFβ (5ng/ml). Noteworthy, the suppressive effect of TGFβ do not only last for short-term incubation times (< 24h) as it is demonstrated here for an IFN-γ ELISA, but is also obvious for long-term proliferation assays (5–6 days). In conclusion, these data demonstrate that TGFβ is a potent inhibitor of CAR T-cells, regardless of their antigen specificity.
Deletion of TGFβ RII via CRISPR/Cas9 system
We prompted us to assess whether genomic disruption of TGFβRII in CAR T-cells will counteract the aforementioned functional impairment of CAR T-cells in the presence of exogenous TGFβ. To achieve this purpose, we designed a 2-step protocol comprising CRISPR/Cas9 based gene knock out of the TGFβRII locus and subsequent CAR IVT-RNA electroporation into those genomically edited primary human T-cells (Fig. 3A). The RNA-guided Cas9 nuclease from the microbial CRISPR adaptive immune system is an efficient genome editing tool which consists of nuclease Cas9 and a single-guide RNA (sgRNA) targeting a 20-bp region of the genomic region of interest [18]. We used the Synthego online tool to design sgRNAs and selected three gRNAs from the top ranked suggested sgRNAs on both strands with at least 50 nucleotides distances (Fig. 3B). The Synthego online tool uses the Doench et al [19] scoring algorithm to select efficient sgRNAs and also ranks the sgRNAs based on the numbers of genomic off targets. Number of potential off target sites for one, two and three mismatches was zero for gRNAs 3/7, while in the case of gRNA5, four off targets emerged with three mismatches. To combine the knock out procedure and CAR expression, briefly, OKT3 activated T-cells were electroporated with Cas9 mRNA alone (Cas9 control), or Cas9 and chemically synthesized O-methyl protected sgRNA targeting TGFβ RII exon 2. Five days later, TGFβ RII ablation was checked using genomic analysis techniques by means of a T7 endonuclease I assay and Sanger sequencing. Cells were subsequently electroporated with saturating amounts of IVT mRNA encoding the anti-MSLN or anti-CLDN6 CAR (Fig. 3A).
To determine the knock out efficiency, genomic DNA was extracted from the cells (Cas9 only and Cas9 with gRNAs 3, 5 or 7) and was submitted to PCR amplification using PCR primers flanking the sgRNA targeting region. An aliquot of the PCR products was subjected to T7 endonuclease I restriction and then run on an agarose gel (Fig. 3c).
T7 endonuclease I recognizes and cleaves mis-matched DNA, which could lead to extra bands on the gel in case of indel formations. The PCR product from control cells and the three gRNAs treated cells were submitted to digestion by T7E1 enzyme. While there were no extra bands in the control, all gRNA treated groups exhibited extra bands which indicates indel formations (Fig. 3C). As outlined in Fig. 3B, each gRNA targets nearby but different genomic sequences of the TGFβRII gene and hence, taking into account to use identical PCR primers, the restriction pattern of T7E1 ended up with variably sized restriction products for each gRNA visible on the gel. The PCR products were also delivered for Sanger sequencing and the data were analyzed for indel frequencies applying a decomposition technique. Base ambiguities commence after base 4 of gRNA7 (5’-GTGA-A/C…), which is consistent with the postulated genomic cut site of Cas9 (Fig. 3D). Again, results confirmed indel formations in all gRNA treated groups (Fig. 3D/E). Although gRNA3 was prone to form less indels in comparison to the two other gRNAs (Fig. 3E), we did not exclude it from our further experiments so as to check if even a lower level of genomic TGFβRII knock out might also have a measurable effect on CAR T-cell functions. In summary, these data exhibited the successful generation of insertions/deletions (INDELs) in the TGFβ RII gene locus. We were able to demonstrate gene ablation to similar degrees for the 3 scrutinized gRNAs in both, CD8 + and CD4 + cells, respectively (Fig. 3E).
TGFβ RII disruption in CAR T-cells enhances their in vitro effector functions
To determine the effect of TGFβRII deficiency on CAR T-cell function, we assessed TGFβRII KO CAR MSLN and CAR CLDN6 T-cells in different assays in vitro. Due to limited KO efficacy, these experiments were done on bulk populations that contained both edited and non-edited cells. As an essential prerequisite for comparison of KO CAR T-cells with WT CAR T-cells, gene ablation of TGFβRII must not interfere with IVT-RNA - mediated CAR expression in T-cells. Indeed, we observed very similar frequencies of CAR expression on T-cells (Fig. 4A): 50% CAR expression could be observed for both, CAR-MSLN electroporated T-cells and TGFβRII KO CAR-MSLN T-cells. Similar to that, we also found up to 70 % CAR expression in electroporated T-cells for CAR CLDN6 irrespective of the genomic editing step. Additionally, we analyzed proliferation of WT or KO CAR T-cells following electroporation in the absence of exogenous TGFβ to determine whether deletion of the TGFβRII gene has any impact on proliferation and/or survival of CAR T-cells. In most tests, we did not monitor any marked alterations in proliferation between WT and TGFβRII KO CAR T-cells and analysis among multiple donors did not result in a statistically elevated difference between WT and KO groups when the latter group was averaged over the 3 different gRNAs (white bars in Fig. 4B, and data not shown). Antigen independent expansion or cytokine secretion of KO CAR T-cells was absent leading to the conclusion that deletion of the TGFβRII does not result in immortality (black bars in Fig. 4B and 5 ).
Again, TGFβ induced reduction of cell proliferation by more than a factor of 2 in WT CAR T-cells. Intriguingly, proliferation rates were restored in CD8 + and CD4 + TGFβRII KO CAR MSLN or CAR CLDN6 T-cells following stimulation with antigen presenting iDCs even in the presence of escalating doses of exogenous TGFβ (Fig. 4B). Of note, for TGFβRII KO CAR MSLN T-cells we still observed a small but steady decline of dose rate - dependent proliferation for 2 out of 3 gRNAs tested. This may result from a higher fraction of non-edited CAR T-cells in bulk CD8 + T-cells used in the assay here, the former which is still susceptible to TGFβ. We also assessed the cytolytic potential of KO TGFβRII CAR T-cells specific for MSLN or CLDN6 against antigen loaded iDCs (Fig. 4C) in a bioluminescence cytolysis assay. However, in contrast to the proliferation and cytokine secretion results discussed before the killing efficiency of these KO CAR T-cells is not affected compared with the WT control.
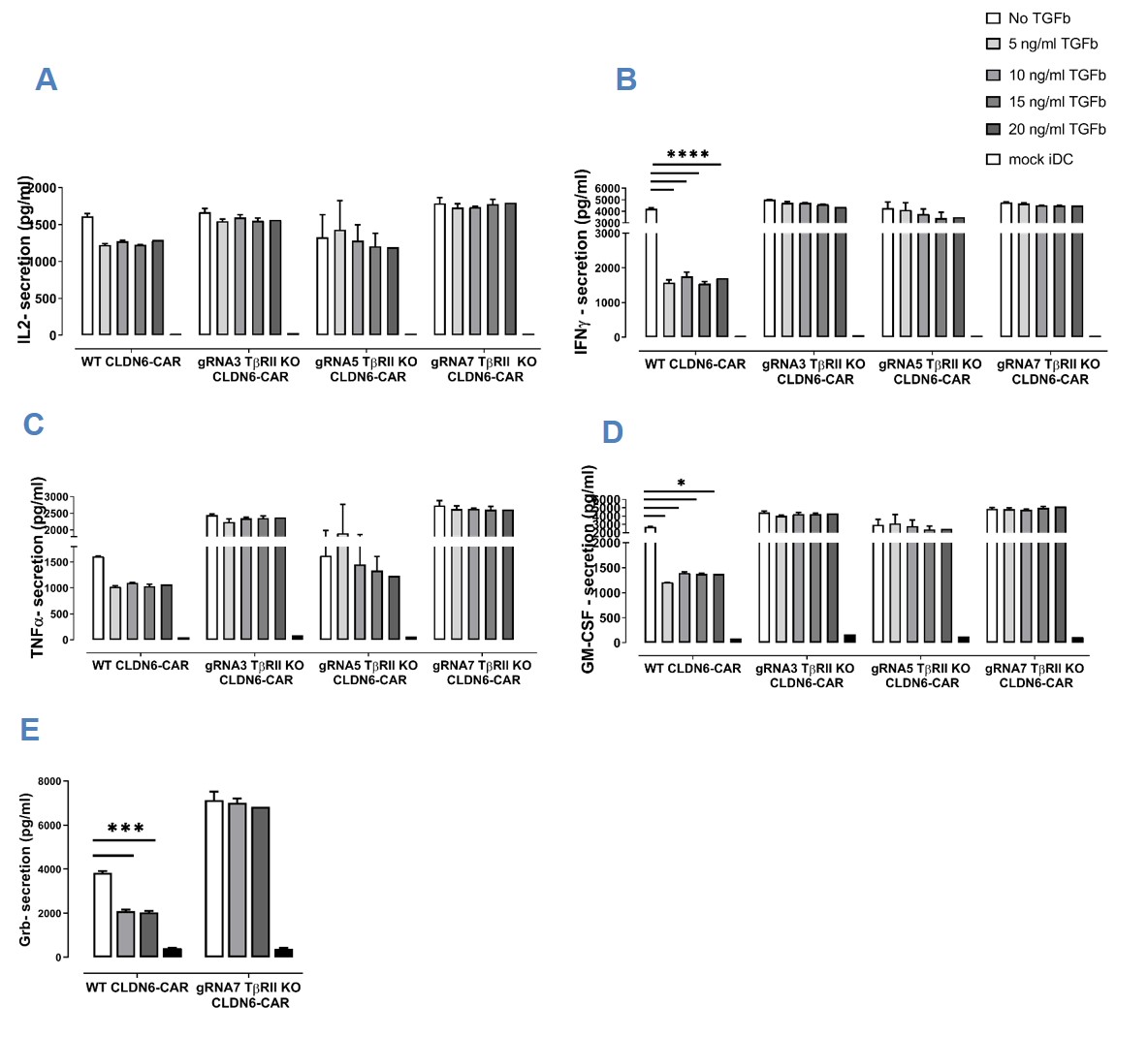
Nonetheless, when compared to WT CAR-T-cells, TGFβRII KO CAR-T-cells released equal amounts of IL-2 (Fig. 5A, S1A), IFN-γ (Fig. 5B, S1B), TNFα (Fig. 5C, S1C), GM-CSF (Fig. 5D, S1D)-cytokines, and GranB (Fig. 5E,S1E) (tested just for one KO group/gRNA7) when they were cocultured with iDCs expressing the cognate antigen in the absence of exogenous TGFβ. This cytokine release was dependent on CAR expression and non-electroporated T-cells (mock) failed to release cytokines in the presence of antigen positive cells (data not shown). Importantly, WT CAR-T-cells lost their ability to secrete cytokines in the presence of exogenous, TGFβ to varying extents (Fig. 5) in the order IFN-γ > GM-CSF > GranB > TNF-α > IL-2 in a TGFβ dose-independent manner. Contrary to that, TGFβRII KO CAR MSLN or CAR CLDN6 T-cells reset the immunusuppressive effect of TGFβ on cytokine secretion almost completely irrespective of the amount of suppression observed for WT CAR T-cells. Secondly, the antagonizing effect of TGFβRII KO CAR MSLN or CAR CLDN6 T-cells was on the whole independent of the dose of exogenous TGFβ added. We postulate that in general, a larger fraction of edited CAR T-cells being present in the bulk population dominate the fraction of non-edited CAR T-cells in the net outcome of function. This means to expect rather a gradual decrease of cytokine secretion in TGFβRII KO CAR T-cells, which is true for most sample groups. However, a steady TGFβ dose rate-dependent increase of GM-CSF and TNF-α secretion became only evident for KO TGFβRII CAR MSLN T-cells. This may be here due to the ratio of edited versus non-edited T-cells, the sensitivity of these cytokine secretion pathways towards TGFβ, and the somewhat lower efficacy of CAR MSLN compared with CAR CLDN6.
In total, these results suggest that TGFβ RII disruption can antagonize in all three KO groups diverse long-term and short-term functional defects induced in WT MSLN or CLDN6 4-1BBζ CAR T-cells treated with exogenous TGFβ. However, our experiments did not reveal any effect of exogenous TGFβ in short term killing assays (4 hours) against immunostimulatory target cells such as iDCs, neither for WT nor KO CAR T-cells.
TGFβ receptor II disruption augments long term cytotoxicity
Several reports showed an inhibitory effect of TGFβ on the cytotoxicity of T-cells and CAR T-cells cells [20–23]. However, we could not reproduce any suppression in short term cytotoxicity assays against iDCs. Hence, we addressed the question whether on long term exposure, TGFβ can hinder the killing function of the CAR T-cells taking into account in particular the chronic prevalence of a suppressive milieu in TME over time in patients. For this, we switched to stably CAR modified T-cells to circumvent transient expression of CARs based on IVT-RNA. Hence, we decided to use virally transduced T-cells and target cells (Fig. 6A). Transduced T-cells were then evaluated for CAR expression (Fig. 6B), viability and TGFβRII KO efficiency. Cytotoxicity was measured using the impedance based technique on an Xcelligence device using adherent CLDN6 + PA-1 Sc12 cell line as target and WT and KO TGFβRII CAR CLDN6- T-cells as effectors. Cells were incubated at an E:T ratio of 5:1.
WT and KO CAR CLDN6 T-cells were not meaningfully different in their cytotoxic function in the absence of TGFβ (Fig. 6C, red curve). However, in the presence of TGFβ, the killing efficacy of WT CAR CLDN6 T-cells was significantly impaired over the entire time range (72h, green curve) to a steady-state level. Remarkably, although all three KO CAR CLDN6 T-cells revealed decreased cytotoxicity to the same extent within the first five hours after coculture setup with target cells, their cytolysis potency recovered in the presence of TGFβ and aligned with the level of cytolysis monitored over time for the non-TGFβ-treated controls (Fig. 6C/D). The initial reduction could be attributed to an immediate suppressive effect of TGFβ captured here in real-time for bulk WT/KO T-cells against immunodeficient tumor cells in contrast to the results shown for immunostimulatory iDCs (Fig. 4C). After this lag-phase TGFβRII KO CAR T-cells became resistant to the TGFβ effect and caught up with non-TGFβ treated CAR T-cells in their cytolytic efficacy. Hence, we postulate that CAR T-cells can indeed be suppressed by TGFβ also in their cytotoxic function as resolved here in a real-time setting. Importantly, this can be reverted with TGFβRII KO CAR T-cells in the long run.
Genomically edited TGFβ unresponsive CAR T-cells can escape control by induced regulatory T-cells.
TGFβ not only commits naïve CD4 cells to differentiate into Tregs, but this cytokine also represents one of the principle mechanisms by which Tregs suppress their primary targeT-cells, namely effector T-cells [24–26]. It has been shown in the past that T-cells which do not respond to TGFβ are able to escape control by regulatory T-cells [27]. It is well known that Tregs are highly abundant in TME, especially in tumors with high level of TGFβ secretion, so we prompted us to verify whether TGFβRII KO CAR T-cells were not only able to withstand TGFβ but also to resist the suppressive effect of Tregs (Fig. 7A). For such experiments, we were challenged to generate Tregs ex vivo from PBMCs in sufficient numbers which were referred to as induced regulatory T-cells (iTregs). The detailed protocol for generating iTregs has been described elsewhere [28]. In brief, CD4 + cells were activated with anti CD3, anti CD28 and IL2. The CD4 + cells which were treated this way represent activated CD4 + T-cells as negative control for immunosuppression. For differentiation into iTregs, rapamycin, retinoic acid and TGFβ were subsequently added to the culture medium. T-cells were activated for up to 6 days, and then analyzed for FOXP3 and CD25 expression to confirm their iTreg phenotype (Fig. 7B). One day before setting up the coculture WT and TGFβRII KO T-cells were electroporated with CAR MSLN (Fig. 7C) or CLDN6 (Fig. 7D) IVT-RNAs and iDCs were electroporated with the respective antigen encoding IVT-RNAs. Since we aimed at demonstrating cessation of the suppressive effect of induced regulatory effector T-cells in a proliferation assay, responder CD8+ T-cells were stained with CFSE before seeding. For setting up the coculture, fixed numbers of iDCs and WT or KO CAR T-cells were coseeded and iTregs were added for different ratios to CAR T-cells (2:1, 1:1 and 0.5:1; Fig. 7C/D). The responder groups comprised beside the WT CAR T-cells as reference 3 different TGFβRII KO CAR Tcell groups each treated with one of the CRISPR/Cas9 gRNAs 3, 5, or 7, respectively. As positive control we included the addition of exogenous TGFβ in the absence of iTregs, to confirm on the one hand that WT CAR T-cells are susceptible to suppression and on the other hand that TGFβRII KO CAR T-cells generated ex vivo are resistant to the anti-proliferative effect of exogenous TGFβ (Fig. 7C/D, second outmost left). Eventually, WT and KO CAR T-cells elicited comparable rates of proliferation in the absence of exogenous TGFβ (Fig. 7C&D outmost left). Activated CD4 + T-cells turned out to be not able to suppress WT and TGFβRII KO CAR T-cells. Intriguingly, in vitro generated iTregs can inhibit proliferation of WT CAR T-cells even at the lowest iTreg to CAR T-cell ratio, ranging from excess 2:1 down to 0.5:1. Most importantly, although iTregs can also inhibit the TGFβRII KO CAR T-cells a little, the suppressive effect is less pronounced in a iTreg dose-dependent fashion compared to that in case of WT CAR T-cell. The remaining suppressive effect in 3 gRNA treated groups may at least in part be explained by the bulk T-cell populations used here, comprising unedited and hence TGFβ-responsive WT CAR T-cells and TGFβRII KO CAR T-cells in each group. The suppressive effect is most likely due to TGFβ produced endogenously by the induced regulatory T-cells and secreted for a paracrine effect because we solely targeted TGFβRII via CRISPR/Cas9 in responder T-cells. Since iTregs have not been equipped with CLDN6 or MSLN-specific CARs, iTreg/iDC*Ag cell/cell contacts are highly unlikely to occur and to contribute to the suppressive effect towards responder T-cells observed here [29].
TGFβRII KO rescues CAR T-cell exhaustion induced by TGFβ
Finally, we explored if TGFβ is also able to foster CAR T-cell exhaustion characterized by decreasing functional responsiveness, using a repetitive antigen stimulation assay, and whether the KO of TGFβRII on CAR T-cells are able to prevent this counter-regulatory effect. In a series of experiments, we took advantage of the impedance based cell killing assay: For this, stably modified CAR T-cells by means of retroviral transduction were a prerequisite in long term restimulation experiments. CAR T-cells were cocultured with the cell line OCVAR3 which expresses CLDN6 endogenously at an E:T ratio of 5:1.
In the first (Fig. 8 Ai) and second (data not shown) round of stimulation TGFβ revealed initial suppression within a few hours (Fig. 8 Aii/Aiii) on cytotoxicity, but was not able to mount a long-lasting suppressive effect on the cytotoxicity of the CAR T-cells, neither on WT nor TGFβRII KO CAR T-cells, the latter shown for gRNA 5 and 7, respectively. The reduction in cytotoxicity in TGFβ-treated samples was roughly the same for WT and TGFβRII KO CAR T-cells. Between 8 and 16 hours of coculture lysis became complete for both groups. In the course of the third round of coculture (Fig. 8 Bi), TGFβ-untreated WT and KO T-cell groups sustained cytolytic function over time. On the other hand, WT CAR T-cells turned out to be for the first time vastly impaired in their cytotoxic function in the presence of TGFβ over the whole time frame (Fig. 8 Bii): 48 hours after setting up the assay lysis dropped down to less than 50% in comparison to the TGFβ untreated group.
In contrast, TGFβ-treated TGFβRII KO CAR T-cells recovered from a short suppression phase within 16 hours and retained full lysis potency comparable to their related TGFβ-untreated groups lasting till the end of experiment (48 h).
We reasoned to see a suppressive effect of TGFβ on WT CAR T-cells only in round 3 of stimulation because the moderate endogenous expression rate of CLDN6 on ovarian cancer c.l. OVCAR3 prevents premature exhaustion of WT CAR T-cells. This contrasts to the much higher expression seen on teratoma c.l. PA1, the latter which promotes early differentiation into senescent effector T-cells and hence, sensitivity towards TGFβ of WT CAR T-cells already after initial antigen-specific stimulation as outlined in Fig. 6.
To sum up the restimulation experiment, TGFβ exerts a less pronounced prohibitive effect on freshly activated CAR T-cells irrespective of being genomically silenced for TGFβRII expression or not, but affects significantly cytotoxicity of repetitively antigen-experienced, and hence senescent CAR T-cells. Knock out of the TGFβRII via CRISPR/Cas9 turns CAR T-cells, after a short lag-phase, into effector T-cells unresponsive towards TGFβ in the long term and emphasizes this approach in generating more potent long-lived CAR T-cells in immunosuppressive TME.


{kind=link}