Aphid population statistics and leaf morphological changes after inoculation with M. rosivorum on R. longicuspis
To understand the reproduction of aphids, we recorded the number of aphids at 0 d, 1 d, 3 d, 5 d and 7 d. The results showed that the number of aphids increased exponentially and after inoculation for 3 d, 5 d and 7 d, the number of aphids was 2 times, 3.3 times and 3.8 times that of the first inoculation, respectively (Fig. 1). These results indicate that M. rosivorum has high reproducibility and ultimately harms the growth conditions of R. longicuspis. According to the changes in the aphid population after aphid inoculation, we selected the samples at 0 d, 3 d and 5 d as the research materials and analysed the RNA-seq, qPCR and metabonomics to explore the early mechanism of Rosa plants responding to aphid stress.
Overview of the transcriptomic analysis
To further understand the molecular mechanism underlying the response of R. longicuspis to M. rosivorum, an RNA-seq analysis was performed. A total of approximately 58.22 GB clean reads were generated from nine biological samples, including six infected and three control samples. The average Q20 value of the raw reads was 95.88% and approximately 84% of the reads were mapped to the reference genome sequences obtained by Trinity splicing. In addition, more than 90.68% of the reads were mapped to the exon region of the reference genome (Table S1). Ultimately, the sequence and expression information of 34202 genes was obtained for subsequent analysis. A principal component analysis (PCA) and Pearson correlation coefficient analysis of the samples based on the FPKM values showed that all the biological replicates exhibited similar expression patterns, indicating the high reliability of our sequencing data (Figure S1). Taken together, the sequencing quality was sufficient for further analysis.
Identification of DEGs in R. longicuspis inoculated with M. rosivorum
To obtain a comprehensive view of the gene expression profile associated with the response of R. longicuspis to M. rosivorum, we used DESeq2 to identify the DEGs. Based on the filtering parameters of FDR < 0.05 and |log2FC|>1, the expression levels of 2845 (1186 upregulated, 1659 downregulated), 2627 (886 upregulated, 1741 downregulated) and 466 (178 upregulated, 288 downregulated) genes were found to differ significantly in the CK-vs.-A3 d, CK-vs.-A5 d, and A3 d-vs.-A5 d groups, respectively. Among those genes, 998 and 904 DEGs were expressed in A3 d and A5 d, respectively (Fig. 2). These results indicate that a large number of genes were upregulated and that few new DEGs were expressed in the R. longicuspis-M. rosivorum interaction.
To understand the functions of the DEGs associated with M. rosivorum, those DEGs were annotated using GOsEq. This annotation resulted in three major categories: biological processes, cellular components, and molecular functions. A comparison of the CK at 3 d and 5 d indicated that most of the DEGs were enriched in the ‘external encapsulating structure’, ‘cell periphery’, ‘cell wall’, ‘cytoskeleton-dependent cytokinesis’ and ‘histone lysine methylation’ categories and other terms. A comparison of 3 d with 5 d showed that most of the DEGs were enriched in the ‘response to reactive oxygen species’, ‘response to oxidative stress’, ‘response to oxygen-containing compound’ and other terms (Fig. 3A). To better understand the main pathways activated under M. rosivorum stress, we conducted a Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment analysis of the DEGs. Between the CK and A3 d libraries, 492 DEGs were assigned to 113 KEGG pathways. Between the CK and A5 d libraries, 484 DEGs were assigned to 109 KEGG pathways. Among these pathways, ‘biosynthesis of secondary metabolites’, ‘metabolic pathways’, ‘phenylpropanoid biosynthesis’, ‘fatty acid biosynthesis’, ‘galactose metabolism’, ‘cutin, suberine and wax biosynthesis’, ‘cyanoamino acid metabolism’ and ‘sesquiterpenoid and triterpenoid biosynthesis’ were significantly enriched. Between the A3 d and A5 d libraries, 105 DEGs were assigned to 51 KEGG pathways. Among these pathways, some were related to plant insect resistance pathways associated with the terms ‘plant-pathogen interaction’, ‘starch and sucrose metabolism’, ‘monoterpenoid biosynthesis’ and ‘brassinosteroid biosynthesis’ (Fig. 3B). Taken together, the results showed that under M. rosivorum stress, the antioxidant system, terpenoid synthesis and secondary metabolite biosynthesis of R. longicuspis were activated to reduce aphid damage in R. longicuspis.
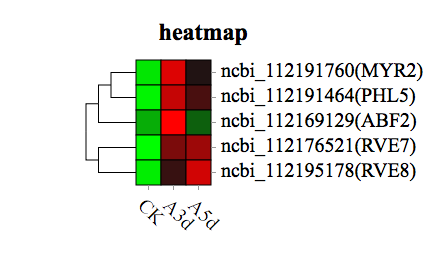
Heatmaps of DEG subclusters were developed to better understand the key DEGs associated with the resistance of R. longicuspis to M. rosivorum. The resulting heatmaps showed the DEGs involved in R. longicuspis-M. rosivorum interactions. Based on their functional annotation, these genes included 1 reactive oxygen species metabolic pathway gene, 3 secondary metabolite synthesis genes, and 13 glutathione metabolism genes, which are shown in the heatmap (Fig. 4). These findings indicated that R. longicuspis responds to M. rosivorum by activating the expression of signal transduction pathway genes, secondary metabolite synthesis genes, antioxidant stress genes and disease resistance genes.
Validation of candidate DEGs based on qPCR analysis
To validate the reliability of the DEGs obtained from the RNA-Seq analyses, the expression levels of 6 candidate genes were analysed using qPCR. These genes included the 1 α-linolenic acid metabolism gene (Rl AOC), 1 starch and sucrose metabolism gene (Rl BGLU11), 2 plant-type cell wall modification genes (Rl EXPB3 and Rl EXPA1), 1 hormone response gene (Rl MBF1C) and 1 homologous recombination gene. The correlation coefficients (r) between the RNA-Seq and qPCR results were calculated for these DEGs (Figure S2; Table S2). The results showed that the correlation coefficients were greater than 0.75, indicating that the RNA-Seq data were reliable.
Overview of the metabonomic Analysis
To understand the changes in metabolites and the possible defence mechanisms of R. longicuspis to infection by M. rosivorum, a metabolite profiling analysis of the R. longicuspis leaf samples (CK, A3 d, A5 d) was performed. A total of 758 metabolites were detected in all samples, and they could be divided into 35 groups (Table S3). The principal component analysis (PCA) showed that the repeatability of different treatments was good. Orthogonal partial least squares discriminant analysis (OPLS-DA) showed that the results of OPLS-DA analysis could be used for subsequent model tests and differential metabolite analysis. Between the CK and A3 d treatments, 31 were upregulated and 34 were downregulated. Between the CK and A5 d treatments, 32 were upregulated and 38 were downregulated. The levels of α-linolenic acid*, γ-linolenic acid*, 2-O-salicyl-6-O-galloyl-D-glucose, scopoletin-7-O-glucuronide, and geniposide increased significantly with the extension of infection time. This finding indicated that these secondary metabolites were involved in the resistance response of R. longicuspis to M. rosivorum.
To identify the main pathways that R. longicuspis uses to respond to M. rosivorum, we mapped the differentially expressed metabolites based on a KEGG biological pathway analysis. A total of 283 metabolites were assigned to 94 KEGG pathways, including ‘metabolic pathways’ (59.72%), ‘biosynthesis of secondary metabolites’ (36.75%), and ‘biosynthesis of antibiotics’ (18.73%), among others (Table S4). Sixty-eight significantly differentially expressed metabolites between the CK and A3 d treatments were assigned to 43 KEGG pathways, including ‘alpha-linolenic acid metabolism’, ‘cyanoamino acid metabolism’, ‘tropane, piperidine and pyridine alkaloid biosynthesis’ and others (Table S5). Seventy significantly differentially expressed metabolites between the CK and A5 d treatments were assigned to 43 KEGG pathways, including ‘aminoacyl-tRNA biosynthesis’, ‘cyanoamino acid metabolism’, ‘glucosinolate biosynthesis’ and others (Table S6). Twenty-six significantly differentially expressed metabolites between the A3 d and A5 d treatments were assigned to 15 KEGG pathways, including ‘alpha-linolenic acid metabolism’ and ‘biosynthesis of unsaturated fatty acids’ (Table S7). The results showed that the metabolic pathways related to disease resistance were significantly enriched, indicating that the defence mechanism of R. longicuspis was activated under M. rosivorum stress.
Glucosinolate biosynthesis
The glucosinolate biosynthesis pathway is involved in plant defence against insects. In our research, 15 metabolites of the glucosinolate biosynthesis pathway exhibited different levels in different treatments (Fig. 5). Among those metabolites, the content levels of L-isoleucine*, L-valine and L-tyrosine were decreased significantly; moreover, compared with the CK, the levels in A3 d and A5 d increased by factors of 1.62, 2.11, and 1.64 and 2.63, 3.06, and 1.64, respectively. In addition, the contents of pyruvic acid, 3-methyl-2-oxobutanoic acid*, (S)-2-hydroxy-2-methyl-3-oxobutanoic acid*, (R)-3-hydroxy-3-methyl-2-oxopentanoic acid* and 2-isopropylmalic acid increased. These results indicate that the glucosinolate biosynthesis pathway may be positively involved in the interaction between R. longicuspis and M. rosivorum.
Glutathione metabolism
Glutathione plays an important role in plant resistance to external stress and reactive oxygen species injury. The ratio of reduced glutathione to oxidized glutathione is one of the important indicators of glutathione activity. In our research, the contents of oxiglutatione and NADP (nicotinamide adenine dinucleotide phosphate) decreased continuously (Fig. 5). Compared with the CK, oxiglutatione decreased by 35.15% and 55.29% in A3 d and A5 d, respectively, and NADP decreased by 12.92% and 17.63% in A3 d and A5 d, respectively. The content level of dehydroascorbic acid first increased and then decreased. Compared with the CK, dehydroascorbic acid increased by 12.42% and 3.86% in A3 d and A5 d, respectively, and L-glutamic acid* increased by 12.08% and 10.84% in A3 d and A5 d, respectively. The results showed that M. rosivorum infection induced the antioxidant system of R. longicuspis and dehydroascorbic acid and GSH were involved in R. longicuspis resistance to oxidative stress caused by M. rosivorum..
Conjoint analysis
A conjoint KEGG enrichment analysis showed 84 comapped pathways, with 35 and 16 comapping pathways between CK-vs.-A3 d and CK-vs.-A5 d and their metabolites, respectively. Interestingly, of these comapped pathways, ‘fatty acid biosynthesis’, ‘metabolic pathways’ and ‘biosynthesis of secondary metabolites’ were significantly enriched in CK-vs.-A3 d, and ‘2-oxocarboxylic acid metabolism’, ‘alpha-linolenic acid metabolism’, ‘sesquiterpenoid and triterpenoid biosynthesis’, ‘linolenic acid metabolism’ and ‘glucosinolate biosynthesis’ were significantly enriched in CK-vs.-A5 d (Fig. 6), indicating that the metabolic pathway related to aphid resistance was activated during aphid stress.
Based on the O2PLS model, the combined analysis of transcriptomics and metabonomic data showed that the model was reliable (R2 > 0.84). The Pearson correlation coefficients showed that the differential expression patterns of DEGs and metabolites were consistent. The correlations between the top 250 DEGs and their metabolites were further selected and are represented as a heat map (Figure S3).
{kind=link}
{kind=link}
{kind=link}