In situ SERS of Cu electrode in the presence of CO2 and NO3−
SERS of the Cu surface in CO2 saturated 0.1 M KHCO3 with 50 mM KNO3 during cyclic voltammetry was performed, and is shown separately for reductive and oxidative scans (Fig. 1b and 1c, respectively). The expected characteristic signals for Cu-oxides were observed in voltametry. In the reductive scan at 0.5 V reduction of CuO to Cu2O is observed, followed by reduction of Cu2O to metallic Cu (Fig. 1a). Those surface chemical changes are in agreement with alterations in intensity of the characteristic peaks at 610 and 520 cm− 1 in the SER spectra, which have been assigned to Cu2O33 (Fig. 1b). At more negative potentials those signals disappear, only to re-appear again in an oxidative scan at 0.5 V (Fig. 1c) and higher, in agreement with the oxidative current in Fig. 1a, which can thus be assigned to oxidation of Cu to Cu2O. Cyclic voltammograms of blank measurements can be found in the Supplementary Figure S1, and Raman spectra in reference conditions in Supplementary Figures S2 (CO2 reduction in bicarbonate solution) and S3 (nitrate reduction in the absence of CO2), confirming that the Raman peaks at 1048 and 1072 cm− 1 in Figs. 1b and 1c can be assigned to nitrate in solution, and surface adsorbed carbonate, respectively.
In a reductive scan (Fig. 1b) up to 0.3 V, little changes are observed in the Raman spectra. When reduction of Cu-oxides is completed, a signal at ~ 2080 cm− 1 appears and grows in intensity at more negative potential. Although the band position is similar to that assigned to adsorbed C ≡ O, the potential at which this band is observed (0.3 V vs RHE) is quite different from previous observations in the absence of nitrate. Moreover, the standard reduction potential of CO2 to CO is -0.106 V vs RHE34, and since the first CV cycle is shown excluding residual CO from previous scans, CO cannot be present at 0.3 V vs RHE. At more negative potentials (-0.7 V vs RHE), the signal at 360 cm− 1 from Cu-CO should appear in case CO is actually formed (Supplementary Figures S2 and S4). However, such signal is not observed at any moment during a CV in the presence of NO3−. In addition, the CO signal at 2080 cm− 1 observed in the absence of nitrate has relatively low intensity compared to the peak observed here (Magnification in Fig. 1b). Surprisingly, although the intensity of adsorbed CO32− at 1070 cm− 1 decreases at more negative potential, it doesn’t disappear completely, and appears more stable in the presence, than in the absence of nitrate (compare the measurement in the absence of nitrate in Supplementary Figure S4). This could suggest that the presence of NO3− in the system prevents conversion of surface adsorbed CO2 to CO to some extent, which is in agreement with the absence of signals of CO in the Raman spectra. The presence of small amounts of NOx in CO2 has been previously found to significantly reduce selectivity of CO2RR (favouring nitrate reduction products), because of the lower onset potential for reduction of NOx compared to CO234,35.
During the oxidative scan (Fig. 1c), the 2080 cm− 1 peak does not disappear at -0.4 V vs RHE like in the case of CO formed by reduction of CO2. This is additional proof that the observed signal originates from a product formed only in the presence of NO3− and CO32−. Finally, when Cu2O is formed (0.5 V vs RHE), the ~ 2080 cm− 1 peak disappears while a new signal at 2150 cm− 1 appears. The Stark effect (Supplementary Figure S5), proving that the signals originate from surface-absorbed species, is observed for both the ~ 2080 cm− 1 and 2150 cm− 1 bands, respectively. Besides CO, carbon-nitrogen triple bonds (C ≡ N) show significant intensity in the wavenumber range of interest, out of which cyanide is the most simple compound43,44. Although it was also reported that Cu-H (adsorbed hydrogen) can appear at the same position in SEIRAS45, in a control experiment in Ar saturated 0.1 M KClO4 signals were absent (Supplementary Figure S6).
In order to prove that the reaction is electrochemically driven, SERS of a Cu surface in CO2 saturated 0.1 M KHCO3 with 50 mM KNO3 was recorded at OCV (Fig. 2a). Clearly bands in the 2000–2500 cm− 1 remain absent as a function of time. At -0.3 V vs RHE, a broad peak at 2080 cm− 1 appears and remains visible throughout the measurement. Subsequently, at OCV when Cu2O signals re-appear due to surface oxidation of metallic Cu, the band at 2150 cm− 1 appears and slightly decreases in intensity over time while shifting position from 2154 cm− 1 to 2149 cm− 1. This suggests that the product formed under reductive potential, changes frequency when oxidation of Cu occurs to form a Cu2O containing surface. Moreover, in order to exclude any homogeneous reactions induced by possible Cu ions present in the solution, cyclic voltammetry in blank electrolytes was performed and SERS at OCV was measured while adding KHCO3 or KNO3 (Fig. 2b). The signal at 2150 cm− 1 was not observed in any experiment, proving that the reaction is driven electrochemically and can be assigned to CuC ≡ N.
A summary of the observed SERS signals can be found in Table 1.
Table 1
Summary of SERS signals observed in this work. *Signals related to cyanide observed in this work.
| Position [cm− 1] | Assignment | Ref. |
| 275 | Cu-CO | 27,36 |
| 360 |
| 350 | CO32− (C-O)asym | 27 |
| 1540 |
| 520 | Cu2O | 33 |
| 610 |
| 1048 | NO3− | 37,38 |
| 1072 | CO32− (C-O)sym | 27,39 |
| 1360 | bicarbonate | 27 |
| 1640 | H2O | 27 |
| 2070 | C ≡ O | 27,39 |
| ~ 2080* | Cu(C ≡ N)n(n−1) | 40,41 |
| ~ 2150* | CuC ≡ N | 40 |
| 300* | Cu-CN | 42 |
We will now discuss if C ≡ N is a possible product of reduction of urea, or formed in a route via intermediates.
Urea decomposition – Does it form Cu-CN?
Surprisingly, the bands observed in the Raman spectra of Fig. 2, do not provide much information regarding (intermediates in) the formation of urea. To evaluate urea chemistry over Cu surfaces, potential dependent SERS analysis of 50 mM urea, in CO2 saturated 0.1 M KHCO3, is shown in Supplementary Figure S7. Several Raman signals can be assigned to adsorbed urea on Cu surfaces, while a strong vibration at 713 cm− 1 is apparent after reduction of Cu2O at 0 V vs RHE (Figure S7a), shifting to lower frequency as a function of increasingly negative potential (Figures S7b and S7c). We speculate this band is due to the C-N vibration (strong at ~ 1005 cm− 1 in solution) of urea, shifted to lower wavenumber by strong adsorption/interaction of urea with the Cu/Cu+ surface. Interestingly, when urea is exposed to Cu at reductive potentials, a Cu-C ≡ N related signal appears in the Raman spectra, as well as the Cu-CO band at ~ 360 cm− 1 at the most negative potentials (< -0.9 V) applied, the result of conversion of CO2 to CO and coinciding with the disappearance of the peak at ~ 710 cm− 1. Apparently urea does not prevent reduction of CO2, contrary to nitrate.
Urea in solution can undergo multiple reactions, but formation of C ≡ N has not been previously reported in reductive conditions46,47. It can hydrolyse to cyanate and ammonium, which is a reversible reaction48. In neutral solution, cyanate may decompose further to produce ammonium ions and carbonate 49. Is should be noted that urea was not formed by reaction of ammonium carbonate solution, suggesting that consecutive cyanate decomposition renders urea decomposition irreversible50. Reduction of cyanate to C ≡ N appears feasible, but was not experimentally proven.
Cu-CN: intermediate to Urea?
Also the role of C ≡ N in the electrochemical path towards urea, if any is formed from CO2 and NO3−, remains difficult to assess on the basis of the Raman experiments. Since the characteristic Raman feature at ~ 709 cm− 1 related to urea adsorption is completely absent in Figs. 1b and 1c, C ≡ N is most likely formed by conversion of a short lived (common) intermediate, such as a C-N species.
In order to prove that the observed signals belong to cyanide, and evaluate potential consecutive reactions of C ≡ N, SERS of the Cu surface in CO2 saturated KHCO3 in the presence of KCN was measured as shown in Fig. 3.
In the presence of 10 ppb of KCN, a signal at 2147 cm− 1 appears (Fig. 3a) which matches the peak observed in our experiments. It was proven that SERS can be successfully used for detection of trace amounts of cyanides as demonstrated for Au in the gas phase42 as well as in solution on Ag51 and Cu surfaces, where Cu(I) surfaces can be very effective in detection by forming CuCN52. The position of the peak is similar to the C ≡ N stretching position (frequency) of solid CuCN40 and surprisingly, it changes depending on CN− concentration (Fig. 3b). In excess of CN− in aqueous solution, CuCN can form higher, soluble complexes, generalized as Cu(CN)n(n−1) (n = 2, 3, 4), which structure depends on the CN−/Cu ratio53. At higher CN− concentration, the Raman signals of Cu2O decrease in intensity (Supplementary Figure S8), which proves chemical reaction of CN− to be likely with (surface) copper oxide, forming higher CuCN-complexes54.
In order to verify the behaviour of the C ≡ N signal during a potential sweep, cyclic voltammetry in 0.1 M KHCO3 with addition of 0.1 ppm of KCN was performed (Fig. 4). The obtained results, i.e. strong signals at ~ 2080 cm− 1, are in excellent agreement with the experimental results obtained in CO2 and NO3− containing electrolyte. Note that NO3− was not added in this measurement, thus those signals must originate from C ≡ N. Cu(CN)n(n−1) can give different Raman signals depending on the type of complex and usually more than one is present in the system, which can depend on the concentration of CN− as well as the pH of the solution40,41. In our system, considering continuous production of CN− at negative potentials in a CV cycle as well as a pH change near the electrode surface it is reasonable to assign the set of ~ 2080 cm− 1 peaks to Cu(CN)n(n−1). Formation of HCN is also possible considering the bulk pH of the electrolyte being 8.6, although this is much more favourable at lower pH41.
If higher KCN concentrations were used (Supplementary Figure S9), Cu2O signals were hardly recorded during a CV, which again confirms chemical attack of CN− on Cu(I)-oxide forming Cu-CN complexes. This likely explains the broad peak observed even at positive potentials, and the very small signal at 2170 cm− 1 attributed to CuCN.
Finally, in order to definitively prove formation of cyanide, isotopic labelling experiments with KH13CO3 and K15NO3 were performed (Supplementary Figure S10). The results show peak shifts with both, 13C and 15N, proving involvement of carbon and nitrogen in the species responsible for the observed vibrations at ~ 2080 and ~ 2150 cm− 1.
Consequences of Cu-C ≡ N formation
The electrode surface was analysed after chronoamperometry at -0.3 V vs RHE. The electrochemical surface area was significantly increased indicating surface roughening, and the XRD pattern shows a specific increase of the Cu (220) orientation (Supplementary Figure S11). Moreover, on SEM images surface changes can be observed (Supplementary Figure S12). These results all indicate surface reorganization occurs by C ≡ N formation, and thus surface instability during electrolysis in the presence of CO2 and NO3− is likely. This might have significant consequences for a practical process for formation of urea, but this requires further study.
Reactions of formate and NH4+
In order to reveal what intermediate species might be responsible for reaction towards cyanide, cyclic voltammetry with formate instead of bicarbonate and ammonium salt instead of nitrate were performed (Supplementary Figure S13 and S14). Since at 0.3 V no CO2 reduction products are expected (see standard reduction potentials of CO2 in Supplementary Table S1), formate was chosen as one of the most simple products from CO2 reduction in a 2e− transfer55. On the other hand, NH4+ was chosen as N-source, since this is the most likely product of NO3− reduction on Cu surfaces56. In both cases, no signals related to cyanide were observed.
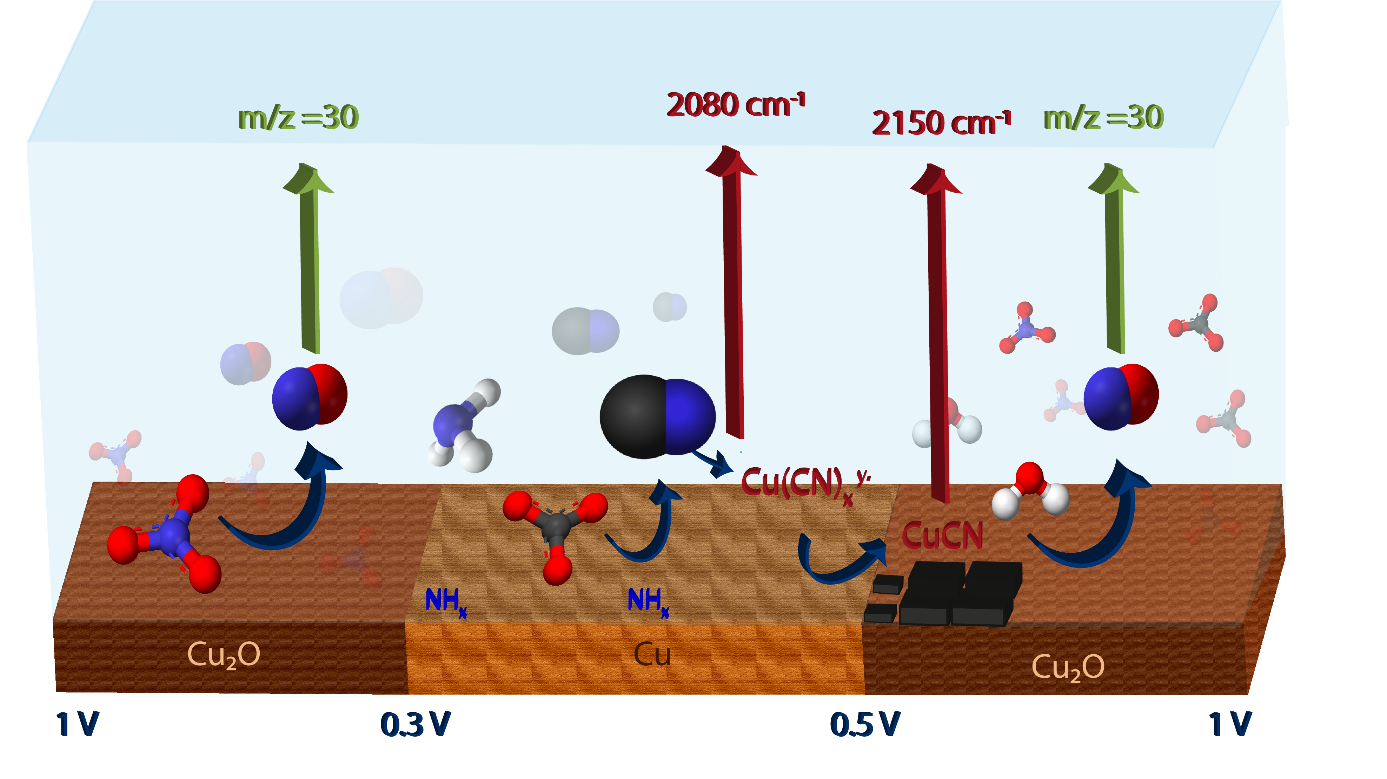
Since cyanide formation requires removal of oxygen from NO3−, it is very likely that NHx intermediates on the electrode surface are involved in the reaction (it was reported that stable NHx adsorbed on cathode surface can exist57). Furthermore, adsorbed carbonate is present in the potential range where adsorbed NHx species exist. Therefore, we conclude that adsorbed NHx on Cu surface reacts with adsorbed CO32− forming cyanide and water, as shown in the schematic overview in Fig. 6.
In situ EC-MS analysis
In order to complement the results obtained by SERS, electrochemical mass spectrometry was used which allows to track gaseous products desorbed from the electrode surface with high sensitivity58. Mass spectra of selected signals recorded during CV scans are shown in Supplementary Figure S15, Figure S16 and Figure S17. Comparison of the m/z: 30 signal, in different electrolytes is shown in Fig. 5a. Besides the obvious difference in m/z: 30 signal, assigned to NO, no other differences were observed. Formation of nitric oxide was confirmed by isotopically labelling experiments, where the peak of m/z: 31 is clearly visible when K15NO3 was used (Supplementary Figure S18).
In CO2 saturated KHCO3 with 50 mM KNO3 as well as in Ar saturated 0.1 M KNO3, NO is observed in the reductive sweep as product of NO3− electroreduction (NO was not found in CO2 saturated KHCO3). The difference in onset potential for NO formation can be explained by different concentrations of NO3− or pH changes in the electrolyte. Although the starting pH is the same (6.8), in the case of CO2 saturated KHCO3 the buffering capacity of the electrolyte renders large pH changes less likely as compared to electrolytes containing KNO3 only. It could also very well be that residual C ≡ N species and adsorbed carbonate inhibit the reduction of NO3− somewhat, further explaining the different trend in NO production as a function of potential. More importantly, nitric oxide is also observed in an oxidative scan if CO2 and NO3− are present. It starts to appear at ~ 0.4 V which matches the onset potential of the formation of the peak at 2150 cm− 1 in SERS experiments. In addition, nitric oxide formation was monitored during chronoamperometry at -0.3 V vs RHE where no signal was detected as shown in Fig. 5b. However, when approaching OCV conditions, the NO signal appears and slowly decreases over time again, which is in agreement with the SERS spectra recorded at OCV (see Fig. 2; blank measurement with 0.1 M KNO3 can be found in Supplementary Figure S19).
The formation of NO during an oxidative scan suggests oxidation of a (surface adsorbed) product, formed in the reductive scan, which based on SERS data is cyanide. Formed cyanide in solution can thus be decomposed by direct electrooxidation59. Cu is effective in this reaction by forming complexes which can oxidize at less positive potentials than free cyanide. It has been previously shown that those complexes can be oxidized at potentials slightly more positive than 0 V vs RHE60. Although the most desired products of oxidation of CN are N2 and CO2; OCN−, CO32−, NO3− and NH3 were also observed46. Moreover, oxidation to NO3− was found to be catalysed by copper oxide61 which very likely involves NO as intermediate. In our analysis system, NO starts to show up at 0.4 V where oxidation of cyanide complexes is possible on Cu electrodes, and the peak of this signal is observed at potentials where copper is oxidized, which is in agreement with literature.
The overall mechanism deduced from SERS and EC-MS experiments is schematically shown in Fig. 6, representing one cyclic voltammetry cycle. This scheme will be discussed in the following summary.
{kind=link}