Asthma is a common chronic pulmonary disease, and its prevalence has been increased over the past few decades [27]. With the increase in morbidity, more interventions and novel tactics are urgently needed for asthma patients to further reduce admission and fatality. Of particular note is the potential role of aging in the parthenogenesis of asthma [28, 29]. Many studies have demonstrated the exist of aging structural cells, immune cells and mesenchymal cells in asthmatic lung although the specific role of aging cells in asthma is still not fully understood [20, 30, 31]. Meanwhile, some related studies have also confirmed the different expression of aging-related genes (such as TP53 and FOXO3) in the development of respiratory diseases [32, 33]. The polymorphism of transcription factor FOXO3 has been shown to be involved in the overactivation of mast cells, down-regulation of anti-inflammatory factors and production of cytokines during the pathogenesis of COPD and asthma [34]. FOXO3 deficiency shows a new role in regulating lung inflammation of COPD/emphysema, which has become a new way to promote the development of pulmonary inflammatory diseases [35]. Similarly, TP53 has been shown to be involved in the progression of COPD by mediating the senescence of multiple lung cells [36]. It has also shown that TP53 is overexpressed in emphysema tissues, which can promote the progression of emphysema in COPD patients [33].
Not only that,as a stable epigenetic marker, DNAm has attracted increasing attention for its involvement in aging-related diseases [37–39]. Aging-related CpG sites have insufficient DNAm or DNA hypermethylation in COPD and other aging-related diseases [40, 41]. Our previous research identified the differential expression and DNAm level of aging-related genes in COPD patients [18]. As asthma and COPD have similar even overlapping clinical phenotypes in chronic inflammation and decreased lung function. In our study, we further explored the methylation change of the previous screened aging-related genes in peripheral venous blood of asthma patients. Indeed, the involvement of these screened 9 aging-related genes in asthma have been extensively studied by previous literatures [42–49]. AREG༌E2F1༌FOXO3༌HDAC1༌MMP2, TGFB1 and TP53 have been verified to be the key molecules through different pathways in asthma [32, 50–56]. Although ATG3 is a key molecule that inducing autophagy damage during aging [57], and NUF2 is a gene closely related to aging of lung cells [58], their specific role in asthma has rarely been studied. The differential expression of ATG3, FOXO3, NUF2 and TP53 in asthma patients were also aligned with former studies [32, 58–60]. In addition, excessive secretion of AREG in the airway after an acute attack of asthma promote airway remodeling [56]. However, the downregulated AREG is present in peripheral blood of elderly asthma patients, which may be due to the discrepancy in different disease processes. It is particularly worth noting that the decreased expression of E2F1 in asthma patients is consistent with what we have previously observed in COPD patients [18]. However, it is different from the expression of E2F1 in the lung tissue of lung cancer patients [60]. One possible reason is the specificity of the sample tissue and pathogenic genes. MMP2, as a member of the matrix metalloproteinase family, has an upward trend in the acute and chronic stages of lung disease. Our results observed the increased expression of MMP2 in asthma patients which is also similar to previous literatures [61].
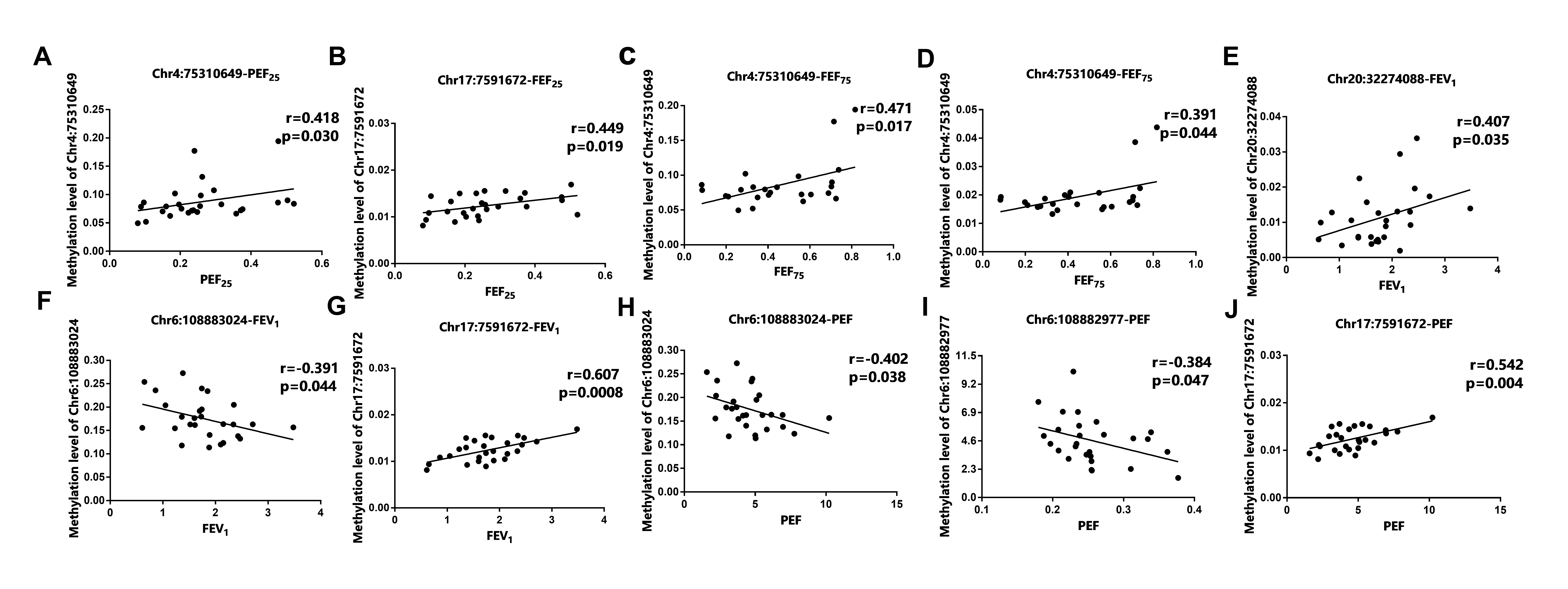
Additionally, we tested the methylation status of the 9 aging-related genes in asthma patients. The methylation level in most DMS of asthma patients were up-regulated, which was consistent with the differential expression of mRNA, indicating that DNAm may be related to the expression of aging-related genes. Moreover, except for ATG3, HDAC1, and TGFB1, correlation analysis showed that the expression of the aging-related genes in peripheral blood of asthma patients was significantly correlated with pulmonary function parameters (FEV1%, FEV1, FVC, PEF, FEF75, FEF50, FEF25). It is known that TGFB1 was a key regulatory cytokine in the process of airway remodeling [62] and HDAC1 played a vital role in the pathogenesis of asthma [63]. This partial difference may be due to the single nucleotide polymorphism in asthma [64]. Chr16:55514392 located in the promoter region has a regulatory effect on gene expression, which is obviously negatively correlated with lung function index (FVC) [65]. Interestingly, Chr16:55514437 is also located at the transcription initiation site, but the specific mechanism of its regulatory genes still needs further study [65]. Furthermore, there were 9 asthma-related CpG sites on the CpG islands of the differential aging-related genes. The ROC curve and PCA analysis of methylation level showed that all the 9 DMSs could be used as potential biomarkers to distinguish asthma from HCs. Most notably, the methylation rate of both single DMS and total 9 DMSs in asthma patients were significantly higher than that of HCs. As the difference in population and ethnicity during the disease process may induce the alteration of methylation, we assumed that the methylation variation rate range can better predict the occurrence of asthma. Our analysis of the 9 DMSs methylation mutation rate also produced a better ROC specificity and sensitivity, suggesting that the DMSs had a great potential to predict asthma from HCs. BALF (IL-25 and IL-33, etc.), induced sputum (eosinophils, Th2 cells, etc.) and airway remodeling (collagen deposition, thickening of basement membrane) could all be used as an useful indicators of asthma diagnosis [66, 67]. However, the detect of DNAm in peripheral blood has greater advantage of widespread access to samples and simple operation. Not only that, DNAm is an important cause of asthma exacerbation, the specific role of allergens and environmental exposure on the epigenetic modification during the development of asthma exacerbation also deserved more attention [68].
Although our study provide potential diagnostic value for asthma assessment, there are also some limitations. Firstly, asthma can be divided into different phenotypes which may have altered epigenetic modification. Besides, our previous work is not comprehensive enough to screen aging-related genes. Moreover, the sample size is relatively small which still need more samples in the future work.
{kind=link}