Selection of the RaPID peptides
N-chloroacetyl-L- or D-Tyr-tRNAfMet was prepared by the use of the respective amino acids esterified with cyanomethyl group (L- or D-ClAc-Tyr-CME) incubated with a flexizyme, eFx 39. Ribosomal synthesis of the macrocyclic peptide library from NNK RNA templates was performed as previously described 40. In brief, 1.2 µM puromycin-linked mRNA library was translated in a methionine deficient FIT reaction containing 25 µM either L- or D- ClAc-Tyr-tRNAfMet for 30 min at 37 ˚C. The reaction was incubated at 25 ˚C for 12 min before disruption of the ribosome-mRNA complex by incubation at 37 ˚C for 30 min in the presence of 20 mM EDTA. The resulting peptide-linked mRNAs were then reverse transcribed using RNase H- reverse transcriptase (Promega) for 1 hour at 42 ˚C and buffer was exchanged for 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.05 vol% Tween-20. Affinity screening was performed by 3 serial passages (counter selections, 10 min each at 4 ˚C) of the library over Covalt-NTA or Streptavidin Dynabeads (Life Technologies), followed by affinity selection against 200 nM protein target immobilized on the same beads for 30 min at 4 ˚C. cDNA was eluted from the beads by heating to 95 ˚C for 5 min, and fractional recovery from the final counter selection (negative control) and the affinity selection step were assessed by quantitative PCR using Sybr Green I on a LightCycler thermal cycler (Roche). Enriched DNA libraries were recovered by PCR and used as input for transcription reactions to generate the mRNA library for the subsequent round of selection.
For high-throughput sequencing, DNA samples from the final round of selection were amplified, purified using a Nucleospin column (Machery-Nagel) and sequenced using a MiSeq high-throughput sequencer (Illumina). Data analysis was performed using CLC sequence viewer 7 software (Qiagen). The target proteins used to derive peptides described in this paper include biotinylated human PlxnB1 ectodomain 6, human MET ectodomain-Fc 5, biotinylated human EGFR ectodomain 41, His-tagged human TrkB ectodomain [Maini, submitted], and biotinylated human a6b1 integrin ectodomain 42.
Cell lines
Cell lines used in this study were obtained from ATCC (HEK293T, MDA-MB-231), RIKEN BRC Cell Bank (Jurkat), Thermo Fisher (Expi293F), and TAKARA bio (AAVpro 293T). Expi293F cells stably expressing human PlxnB1 6 and CHO-K1 cells stably expressing human MET 43 were established previously. All cell lines were routinely tested for the presence of mycoplasma.
Protein design, construction, and expression
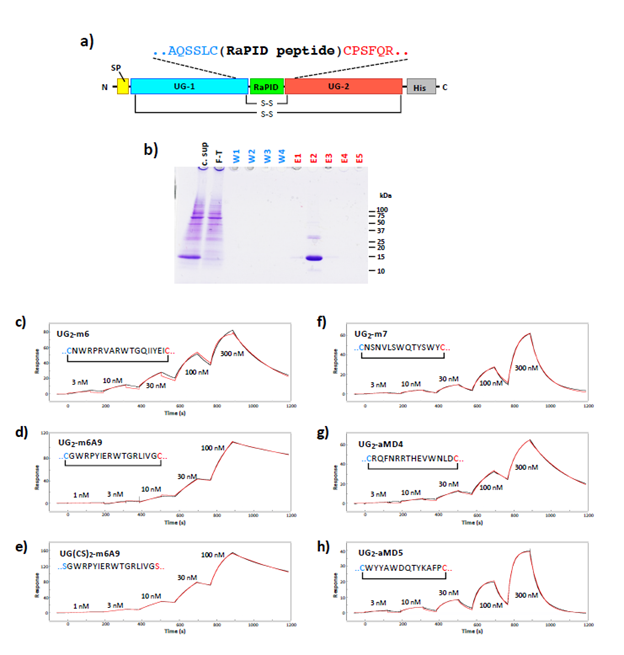
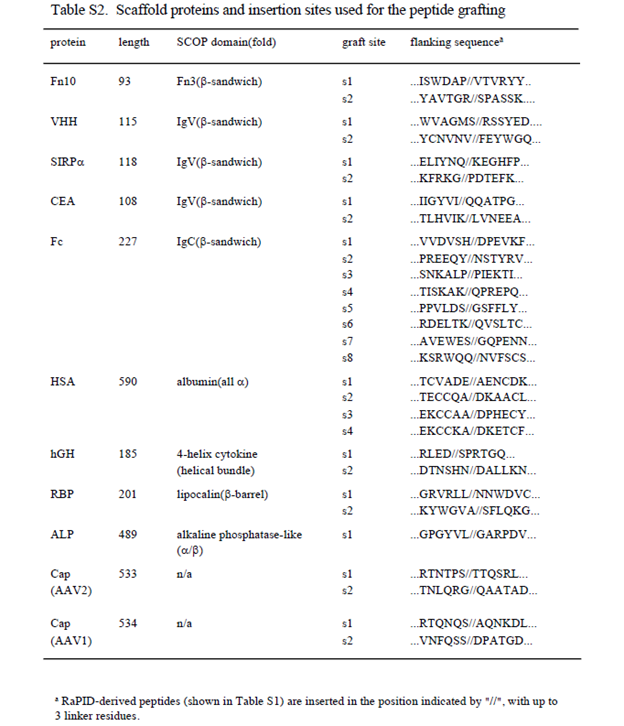
To design peptide-grafted proteins, each scaffold protein was structurally inspected to find appropriate insertion point where the Ca distance between the two anchorpoint residues located at chain ends (for dimer grafting, Fig.1a(i)) or within an exposed loop (for loop grafting, Fig.1a(ii)) were less than 7Å. Internal sequence of the RaPID-derived cyclic peptides (Table S1) were then inserted into these identified sites (Table S2) with up to three spacer residues of either Gly or Ser at both ends. All expression constructs were made using pcDNA3.1-based backbone with appropriate signal peptide and a tag or fusion partner to allow efficient purification and beads-pulldown assay. For the construction of the single-chain UG linked by the peptide (generally called UG2-(peptide name)), two DNA fragments coding for human UG (UniProt P11684) and a peptide-coding region were assembled as shown in Figure S1a by extension PCR. For the construction of Fc-only protein, human IgG1 Fc (residues 104-330, UniProt P01857) was used without any tags. For Fc-fused b-sandwich domains, following regions were amplified from the original cDNAs or synthesized DNAs and fused with the human IgG1 Fc; residues 1539-1631 of human fibronectin (UniProt P02751-15); residues 35-142 of human CEA (UniProt P06731), residue 31-148 of human SIRPa (UniProt P78324), and residue 2-116 of anti-GFP single-domain antibody (PDB ID: 3OGO, 44). Full length HSA (UniProt P02768), hGH (UniProt P01241), RBP (UniProt P02753), and ALP (UniProt P05187) coding regions were cloned in the pcDNA3.1 vectors with C-terminal His tag (HSA , hGHn and ALP) or C-terminal PA tag 45(for RBP). Using these constructs as templates, peptide-inserted variants were prepared by extension PCR, followed by the verification of DNA sequences. For the construction of full-length IgG containing peptide grafts at the Fc region (i.e., addbody), the variable regions of heavy and light chains of YW64.3, avelumab, and OKT3 were gene-synthesized using the publicly available amino acid sequences and formulated in the form of human IgG1/kappa, and the peptide-insertion was performed as described above. For the addbodies containing more than one peptide insertions, the extension PCR process was repeated to incorporate different peptide sequences at different sites. Coding region of all expression constructs were verified by DNA sequencing. Protein expressions were performed using Expi293 expression system (Thermo Fisher) unless otherwise indicated. UG2 proteins were purified from the culture supernatants using Ni-NTA-agarose resin as shown in Fig.S1b, buffer-exchanged to 20 mM Tris, 150 mM NaCl, pH 7.5 (TBS), concentrated to ~1 mg/ml, and stored at -80°C until used.
Beads pulldown
In order to assess the binding ability of various proteins grafted with m6A9 (PlxnB1 binder) or aMD4 (MET binder) in parallel, simple beads-pulldown method was utilized. To this end, soluble ectodomain fragments of human PlxnB1 (residues 1-535) and human MET (residues 1-931) with different tags were expressed and captured onto the beads immobilized with Protein A (for Fc-tagged version), anti-PA tag antibody NZ-1 (for PA-tagged version), or anti-MAP tag 30 antibody PMab-1 (for MAP-tagged version). After brief washing, the beads were further incubated with the culture supernatants containing various peptide-grafted proteins. Bound proteins were then eluted by adding SDS-containing buffer, and analyzed by SDS-PAGE. Binding specificity was confirmed by the lack of nonspecific binding of control scaffold proteins with no peptide insertions.
Kinetic binding measurement using Biacore (SPR)
The ectodomain fragments of human PlxnB1(1-535) or human MET(1-931) were biotinylated via BirA-mediated biosynthetic labeling using the protocol described previously 6, and immobilized onto a Series S sensor chip SA (GE Healthcare) at a surface density of ~930 RU (PlxnB1) and ~840 RU (MET), respectively. The binding was evaluated by injecting peptide-grafted UG solutions serially diluted using the running buffer (20 mM HEPES-NaOH (pH7.5), 150 mM NaCl, 0.05% Surfactant P20). The runs were conducted in a single cycle kinetics mode employing the following parameters; flow rate of 30 µl/min, contact time of 120 s, and dissociation time of 300 s. After each run, the surface was regenerated by injecting the regeneration buffer (10 mM Glycine-HCl (pH 3.0), 1M NaCl) until the response returned to the original baseline level. The binding curves of the measurement cell (immobilized with PlxnB1/MET) were subtracted with that of reference cell (unimmobilized), and used to derive kinetic binding values. Data were obtained using a Biacore T200 instrument (GE Healthcare) at 25°C, and the results were analysed by using Biacore T200 evaluation software version 4.1.
Flow cytometry
To measure binding of peptide-grafted Fc proteins to the respective target receptors, HEK 293T cells were transiently transfected with plasmids coding for various full-length human receptors including PlxnB1, MET, EGFR, TrkB, integrin a6b1, or Nrp1 using X-tremeGENE HP (Merck #6366236001), and detached from dishes by a brief treatment with trypsin/EDTA at 2 days post transfection, followed by an incubation with peptide-grafted Fc proteins diluted at ~10 µg/ml for 1.5 h. After washing twice with PBS, cells were incubated with AlexaFluor 488-labeled goat anti-human IgG (1:400 dilution, Thermo Fisher, A11013) at room temperature for 30 min. To measure binding of peptide-grafted IgG (addbodies), either the HEK293T transient transfectants or cell lines with endogenous expression of PD-L1 (MDA-MB-231), MET (MET-CHO), and CD3 (Jurkat) were used. Stained cells were analyzed on an EC800 system (Sony) and the data were analyzed with FlowJo software (Tomy Digital Biology).
Heterotypic cell-cell attachment assay
The CHO cells stably expressing human MET 43 were labeled by a fluorescent membrane marker DiI (Biotium, #30023) and plated in a 24-well plate (Thermo Fisher, #142475) at 5 × 105 cells/well in F12 growth medium containing 10% FCS. After 5 h, the cells were overlaid with Jurkat cells labeled by fluorescent membrane marker NeuroDiO (Biotium, #30021), which had been preincubated with purified OKT3 or aMD4-grafted OKT3 addbody at 5 µg/ml for 1.5 h on ice, and then further incubated for 30 min at 37 ºC. After removing the non-adherent Jurkat cells by gentle washing with ice-cold PBS for three times, the samples were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, and the phase-contrast and fluorescence cell images were recorded using TRITC filter set (for MET-CHO) and GFP filter set (for Jurkat) for at least 8 field views with a BZ-X700 digital fluorescence microscope (Keyence). The number of CHO cells (red fluorescence) and Jurkat cells (green fluorescence) cells were analyzed by using the hybrid cell count software (Keyence), and the average number of Jurkat cells attached per area were evaluated as the score of addbody-mediated heterotypic cell-cell attachment.
Recombinant production of AAV mutants and gene transduction assays
Recombinant AAV2 vector was prepared using an AAVpro Helper Free System (TAKARA, #6230). As the source of wild-type and mutant Cap subunits to prepare peptide-grafted AAV2 capsid, we modified the pRC2-mi342 plasmid encoding Rep and Cap genes. In order to avoid unwanted mutations introduced in this large (8.2 kb) plasmid, we first introduced unique AgeI and NheI sites flanking s1 and s2 sites to prepare pRC2-mi342AN. Various peptide insertion mutants were made on the ~0.8 kb AgeI-NheI fragment in a separate vector, and swapped into pRC2-mi342AN after sequence verification. As a result, pRC2-mi342AN plasmids with the following three mutations were constructed; PA_s1, PA_s2, and m6A9_s2. For the production of recombinant AAV1 vectors, pAAV2-1 plasmid carrying Rep gene from AAV2 and Cap gene from AAV1 (Addgene, #112862) was used. As in the case of AAV2 construction, mutations (PA_s2 and aMD4_s2) were introduced in the ~0.8 kb BsiWI-SbfI fragment in a separate vector before the final cloning in the pAAV2-1 plasmid. AAVpro-293T cell line (AAV-293, TAKARA, Z2273N) was used for AAV production and was cultured in 10% FCS/DMEM supplemented with 1 % non-essential amino acids (NEAA, Sigma, M7145-100mL) and 0.5 % penicillin/streptomycin (P/S, Sigma, P4458-100ML). AAV-293 cells were seeded at 8.0 × 105 cells/well in 6-well cell culture plate (Thermo Fischer, #24465). About 30 minutes before transfection, the culture medium was changed with 5 % FCS/DMEM containing 1 % NEAA and 0.5% P/S. Cells were transfected with 1 µg/well of a mixture of modified pRC2-mi342 or pAAV2-1 plasmids, 1 µg/well of pHelper plasmid, and 1 µg/well of pAAV-CMV-Fluc or pAAV-CAG-AcGFP plasmid (a kind gift from Takahisa Furukawa, Osaka University). Transfection was performed using 6 µg of PEI-Max (Polysciences, #24765-1). After 24 h post transfection, the culture medium was changed with 1 % FCS/DMEM supplemented with 1 % Glutamax (Gibco, #35050-061), 1 % NEAA and 0.5 % P/S. Seventy two hours after transfection, cells were collected and recombinant AAV was extracted using AAVpro extraction solution (TAKARA, #6235), and concentrated by ultrafiltration in PBS using Amicon ultra (Millipore, 100 kDa cutoff, UFC510024). Virus titers were determined by qPCR analysis of AAV genome copies using AAVpro titration kit ver. 2 (TAKARA, #6233). For the AcGFP gene transduction, Expi293F cells stably expressing human PlxnB1 6 or the parental cells were seeded into each well of 96-well black wall plate (Greiner, #655090) at 5,000 cells/well and were infected with WT or mutant AAV2 at MOI = 6 × 105. Cells were cultured in 5 % FCS/DMEM including 1 % NEAA and 0.5 % P/S. In gene transduction with WT or mutant AAV1, CHO-K1 cells stably expressing human MET 43 or the parental cells were used and cultured in 5 % FCS/Ham’s F12 (Wako, 087-08335) containing 0.5 % P/S. These cells were seeded at 10,000 cells/well and were infected at MOI = 5 × 104. Fluorescent microscope images were recorded after two days of infection by BZ-X700 microscope. For the luciferase gene transduction, cells were plated at 10,000 cells/well into 96-well black wall plate and cultured for one day, followed by infection with mutant AAV1s and AAV2s at MOI = 1.25 × 104, 2.5 × 104 and 5.0 × 104. Cells were cultured for additional 2 days in 5 % FCS/DMEM containing 1 % NEAA and 0.5 % P/S or 5 % FCS/Ham’s F12 containing 0.5 % P/S, and the luciferase activity was determined by measuring luminescence for 0.5 second using Luciferase Assay System (Promega, E1501) in a Glomax NAVIGATOR (Promega, GM2000). Three or four technical replicates for each infection condition were measured.
{kind=link}
{kind=link}
{kind=link}