X-ALD is a demyelinating and neurodegenerative disorder, due to the loss-of-function mutation of ABCD1 gene. ABCD1 gene codes the peroxisome transporter protein ALDP, which plays an important role in regulating peroxisome oxidation and degradation of VLCFAs [5]. The dysfunction of ALDP, a fatty acid transporter, results in the impaired VLCFA oxidation and excessive VLCFAs accumulation in tissues [5, 10]. Excessive VLCFAs in cytoplasm leads to mitochondrion dysfunction and ER stress, finally caused cell death. In addition, accumulated VLCFAs arouses inflammation, results in demyelinating and neurodegeneration.
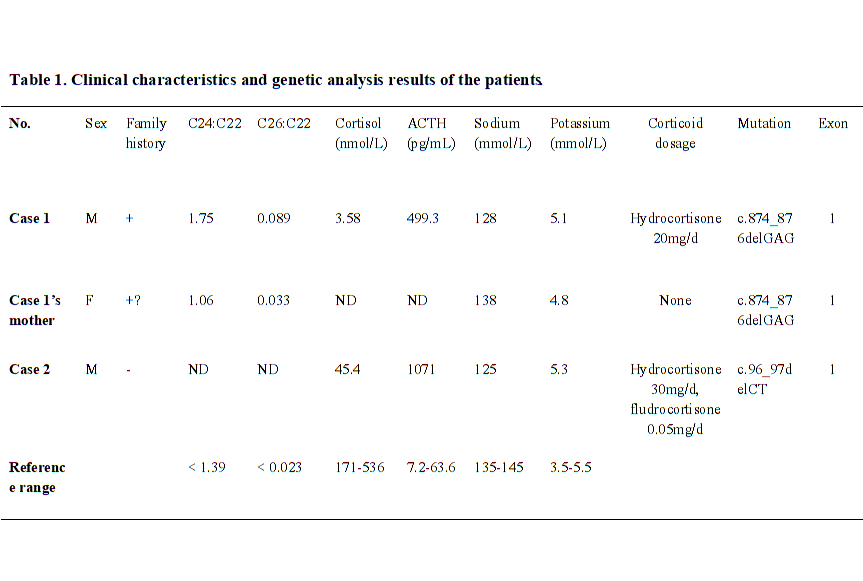
In this study, we reported two Chinese X-ALD pedigree. Both of the probands were diagnosed as adrenal insufficiency at beginning without neuropsychiatric symptoms for decades, but developed into neurological disorders at their late 20’s, and showed rapidly progression and poor prognosis. Two novel mutations in exon 1 of ABCD1 gene was detected and suggested to be associated with the disease.
According to the previous reports, the majority is cerebral ALD among those seven types of ALD [11]. CCALD is early onset with a feature of rapidly progressed neuro-inflammatory of cerebral demyelination. Neurological symptoms of AMN commonly occur from the ages of 20 s, with manifests as a chronic progressive paraparesis, accompanied with sensory and motor disturbances [12]. Due to the variable clinical manifestations, some are difficult to make the diagnose at early stage [13]. Adrenocortical insufficiency was the reported as the first clinical manifestation of 38% X-ALD, greater than the group of spinal cord 25% and cerebral disorders 14.5% [9]. To be noticed, some of those male patients diagnosed as adrenal insufficiency may develop with cerebral or myeloneuropathy ultimately, and show a rapid progressive neurological symptoms and poor prognosis. On the contrast, female ALD patients rarely show adrenocortical insufficiency or cerebral ALD. Most of female patients suffer from lately-onset myelopathy [10, 14]. We reported two cases both were firstly diagnosed as Addison’s disease, and then the therapy focused on the adrenocortical replacement. However, after the neurological symptoms and signs become apparent, both of the cases were rapidly progressed. The therapy including fatty acid restriction or neurotrophic treatment did not show therapeutic effect. Therefore, at diagnosis of congenital adrenal insufficiency, the probability of ALD should be fully considered especially for male patients [8, 15]. To make the early diagnosis, family history, MRI scan of brain or spinal cord, VLCFAs measurement, and genetic screening may be helpful for those patients with subtle early neurological manifestations.
We identified two novel point mutations in exon 1 of ABCD1 gene in the pedigree of first case (c.874_876delGAG, p.Glu292del) and in second case (c.96_97delCT, p.Tyr33Profs*161). The genetic sequencing demonstrated nucleotides deletion, resulting in those frameshift mutations. Based on the clinical symptoms and positive neurologic signs, family history, imaging and biochemical characteristics, we suggest those mutations as pathogenic variants. However, the function of ALDP was not further studied, and the biopsy of brain, spinal cord, and adrenal gland were not done. Based on the elevated plasma VLCFAs, the deleterious change of ALDP impaired the cellular beta-oxidation of fatty acid [16].
There are more than thousand mutations have been reported in ALD database (http://www.x-ald.nl/). Among them, 66.9% of them are pathogenic mutations, 8.93% are synonymous, 3.14% are benign, 18.73% are VUS (variant of undetermined significance), and 1.74% are status unknown mutations detected during the screening of newborns (Fig. 3A). More than 78% are point mutations, other types including 12.37% deletion, 2% del-insertion, 3.92% insertion, and 3.14% duplication (Fig. 3B). Most of the mutations locate in the exon (92.94%), others in the intron (6.18%), 5’UTR (0.61%) and 3’UTR (0.26%), respectively [4]. Though some variations may not affect protein translation or function, they still represent polymorphisms. The genotype-phenotype correlationship have not been identified clearly [17–18]. In the database, nearly half of the mutations were reported anchoring in exon 1, which encodes the transmembrane domain (TMD) of the protein, contains ligand-specific binding sites and plays an important role in the localization of ALDP [19]. Another clustering of mutations occurs in exon 6, which encode the ATP binding domain of ALDP [20]. ALDP locates in the peroxisome membrane, to transport VLCFAs from cytosol into the peroxisome for degradation. Pathogenic ABCD1 mutations may induce defective the stability of ALDP transmembrane structure region and ATPase activity, to affect the localization of VLCFAs to the peroxisome correctly. As a result, VLCFAs cannot be transported into peroxisomes for β-oxidation, then accumulate mainly in the white matter, neuron axons of central nervous system, and adrenal cortex. Excess VLCFAs accumulation leads to destructive progression by triggering oxidative stress and mitochondrial dysfunction, chronic inflammation, lipid-induced neuron apoptosis, and degenerative changes in the nervous system, and finally leading to neurodegeneration and adrenal insufficiency [3, 21].
Therapeutic strategies include adrenocortical replacement, some of the adrenal insufficiency patients may require both glucocorticoid and mineralocorticoid to maintain water electrolyte balance. For severe adrenal insufficiency, or even adrenal crisis, timely initiation of steroid replacement is essential. Nutrition intervention, for instance, low-fat diet and Lorenzo's Oil may reduce VLCFAs level [22]. In this way, the risk of development with cerebral disorders may be reduced. However, they lack the evidence to improve the symptoms and progression. Another efficiency approach is hematopoietic stem cell transplantation (HSCT), especially in the early stage of the disease [23]. Transplantation with autologous bone marrow transfected in vitro with ABCD1gene modification has been performed [24–25]. Bone marrow transplant has been reported in CCALD therapy, most of the patients achieved improved prognosis. However, the pre-existed severe symptoms may influence the outcome after transplantation [26]. In animal experiments, Abcd1− mice show reduction of the mitochondrial biogenesis driven by PGC-1α/PPARγ pathway [27]. Oxidative stress is involved in the physiopathological mechanism of neurodegenerative cascade and mitochondrial depletion [28]. Pioglitazone, a PPARγ/PGC-1α axis activator may modulate the anti-inflammatory and anti-oxidant response. Recently, pioglitazone is under phase II Clinical trials for adrenoleukodystrophy patients, reveals a potential for clinical translation [29]. Therapy options are limited, in addition, some therapeutic approaches relay on the early diagnosis and intervention. Therefore, gene sequencing analysis may be an important tool for newborn screening and genetic counseling.