Along with the rapid development of high-throughput technologies, DNA chips and second-generation sequencing technologies have generated massive amounts of data, and researchers need bioinformatics to process useful information[23]. Bioinformatics is an interdisciplinary subject combining computer and life science, which comprehensively computer science, statistics, biological science and other theories to calculate and analyze the potential significance of extensive biological data[24]. More and more bioinformatics platforms and analysis software have come into being. Their data contains genome information and functional integration, which can significantly improve the extraction and analysis of biological data. Data sharing based on platforms also effectively reduces the experimental and time cost.
Changes in cancer metabolic processes such as glucose metabolism and amino acid metabolism are characteristic of cancer[25]. The characteristics of metabolomics can better understand the pathophysiological changes of HCC and provide great potential for the development of new methods for HCC treatment[26]. For instance, These metabolic pathways (bile-acid biosynthesis, tryptophan metabolism, urea-cycle metabolism, and Citric acid cycle) were significantly changed in HCC group[27]. Previous study indicated that glycolysis and amino acid metabolism had a closely association with the development and progression of HCC by multi-omics analyses[28, 29]. Moreover, abnormal lipid metabolism has also been found in HCC patients[30].
In this study, we studied MRGs and TFs changes in HCC patients by bioinformatics. After identifying X MRGs in the TCGA data sets as capable of identifying key clinicopathological features of HCC, we established risk signatures of MRGs by LASSO regression analysis. We identified the seven-gene prognostic model of HCC by using the formula to calculate the prognostic score: [Y = DHDH*0.658 + ENO1*0.052 + G6PD*0.043 + LPCAT1*0.147 + PDE6D*(-0.154) + PIGU*0.040 + PPAT* 0.604]. According to our risk signature, patients in the high-risk group tend to be associated with poor prognosis, which has a significantly higher expression of DHDH, ENO1, G6PD, LPCAT1, PIGU, PPAT, but lower expression of PDE6D.
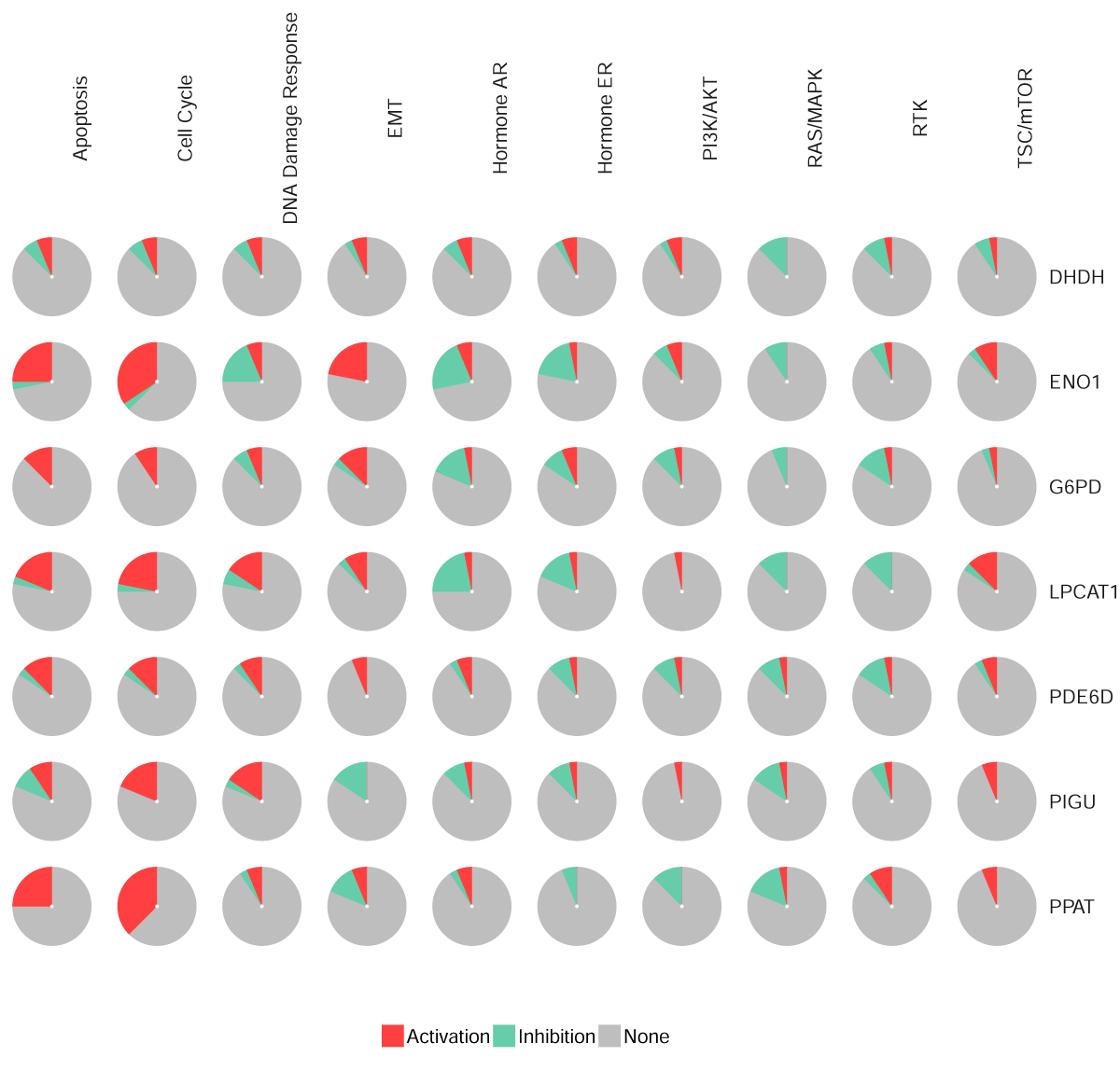
The model signature genes ENO1, G6PD, LPCAT1, PIGU, and PDE6D have been found to be involved in the development and progression of HCC. ENO1 (α-enolase), a key enzyme of glycolysis, can promote the conversion of 2-phosphoglycerate to phosphoenolpyruvate 1, which can enhance the ability of proliferation in HCC[31]. Furthermore, previous studies have shown that ENO1 is upregulated in HCC tissues, which is related to tumor differentiation and progression[32]. G6PD (Glucose-6-phosphate dehydrogenase) is the first enzyme and rate limiting enzyme of pentose phosphate pathway[33]. The expression of G6PD in HCC patients and HCC cell lines are increased, which can promote migration and invasion by epithelial-mesenchymal transition (EMT)[34]. LPCAT1 (lysophosphatidylcholine acyltransferases 1) can acylate the unsaturated acyl group to maintain the integrity of the cell membrane[35], which can increase the ability of cell proliferation, migration and invasion in HCC[36]. PIGU (Phosphatidylinositol glycan anchor biosynthesis class U) plays its carcinogenic role by enhancing GPI-T activity and anchor-binding substrates including urokinase plasminogen activator surface receptors[37], which relate to poor prognosis in HCC, and nomogram-based risk scores that combine PIGU level with the standard TNM tend to be a more powerful set of vehicle for predicting prognosis[38]. Finally, Peter Dietrich and his colleagues found that PDE6D (rod-specific photoreceptor cGMP phosphodiesterase) might affect different cytoplasmic and nuclear pathways in HCC, as well as other types of cancer. PDE6D could promote the ability of proliferation, migration, invasion and sorafenib resistance in HCC cells, so PDE6D's great potential might be a new therapeutic and diagnostic target for HCC progression and chemotherapy resistance.
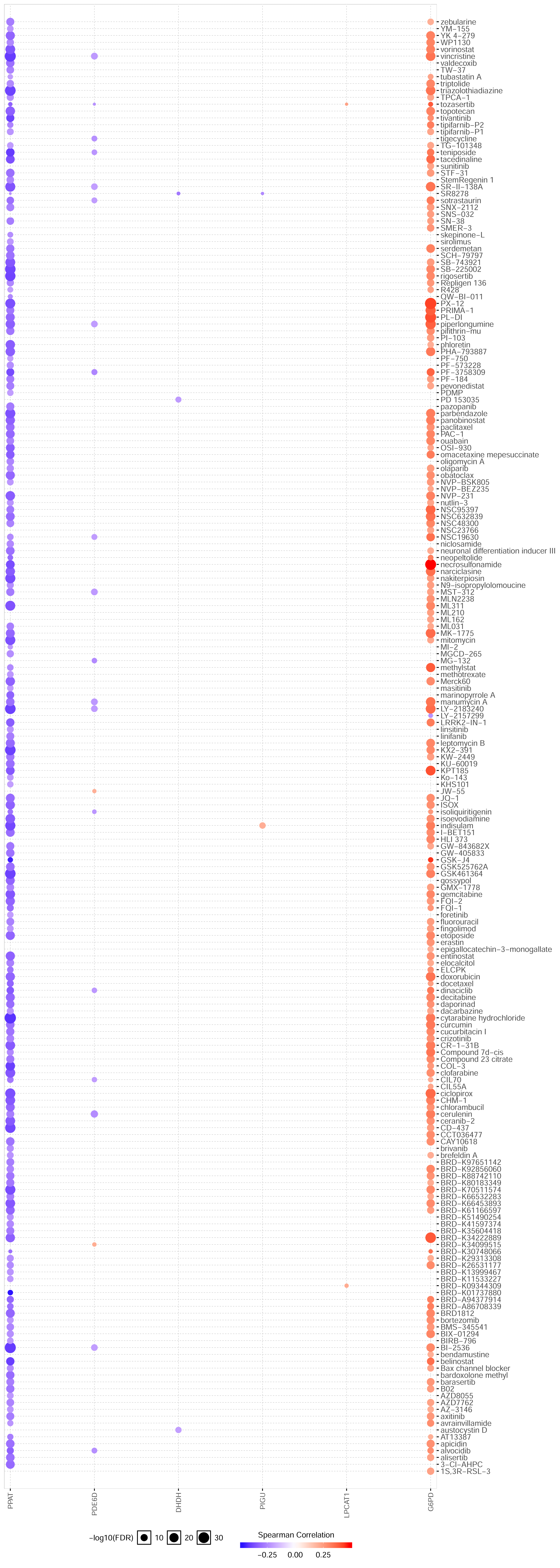
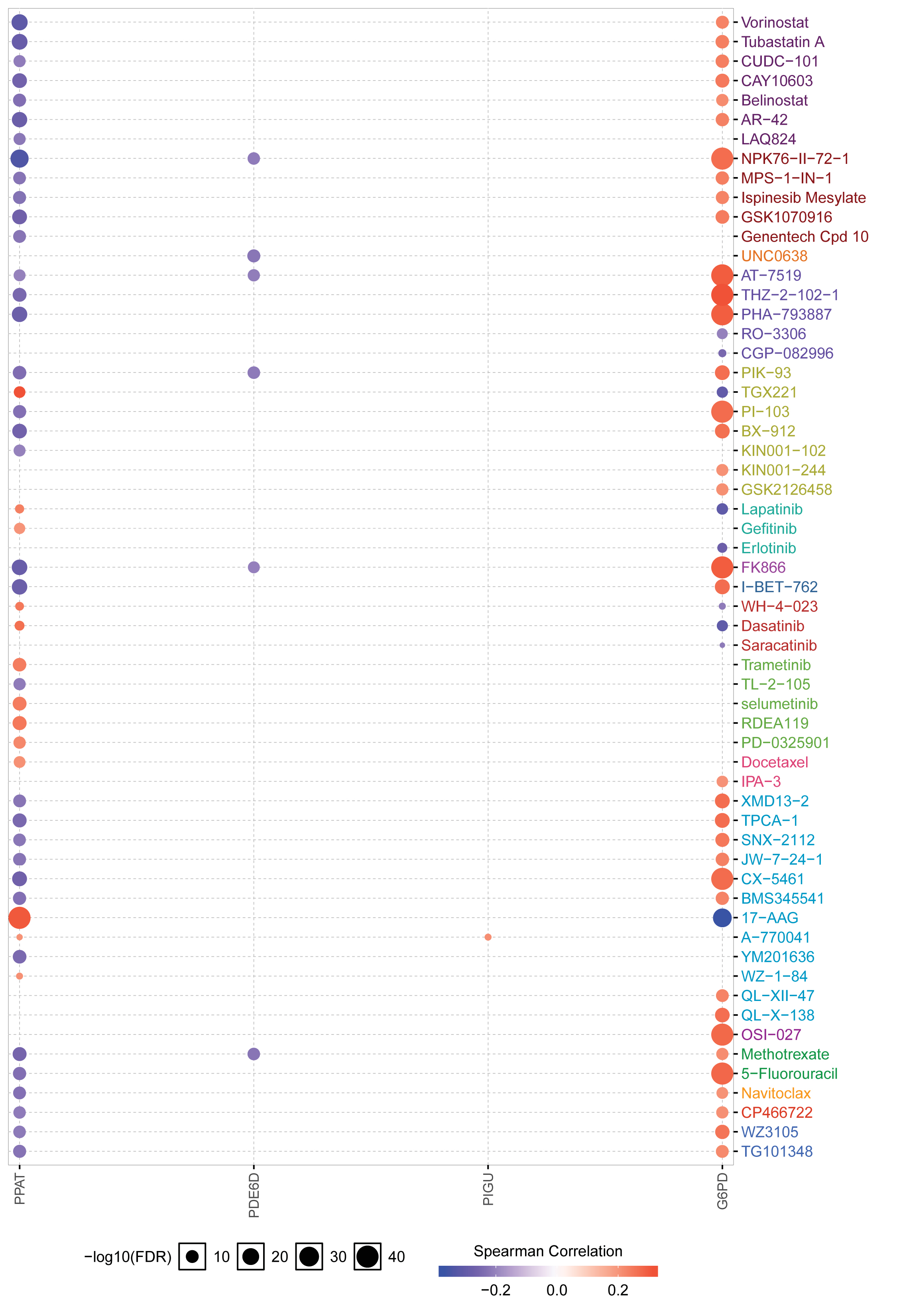
To investigate the potential molecular mechanisms by which gene signatures influence prognosis, GSEA analysis was utilized. The results showed that the gene expression changes in the prognosis model mainly affected monocarboxylic acid catabolic process, organic acid catabolic process, and positive regulation of cell cycle phase transition, providing clues for further research. In KEGG terms of GSEA, cell cycle was the most affected pathophysiological pathway. In summary, these seven hub signatures gene might influence cell cycle to mediate the HCC progression by multiple metabolic pathways. Furthermore, the drug sensitivity analysis based on GSCALite indicated that G6PD might be involved in the resistance of multiple chemotherapy drugs, contributed by apoptosis and cell cycle. However, the relationship between HCC progression and G6PD expression still be not confirmed, and our correlation analysis between IHC staining and clinical pathological parameters might provide some clues to research in this area. Taken together, our result suggested that MRGs, especially in G6PD, might play a role in the development and progression of HCC.
{kind=link}
{kind=link}
{kind=link}