Several SARS-CoV-2 RBD and hACE2 complex structures have been solved and deposited in Protein Data Bank (PDB). We use pdb 6M17 for our analysis. The peptide inhibitor is derived from ABP (pdb code 6H9H) as described in our previous studies49.
Docking analysis:
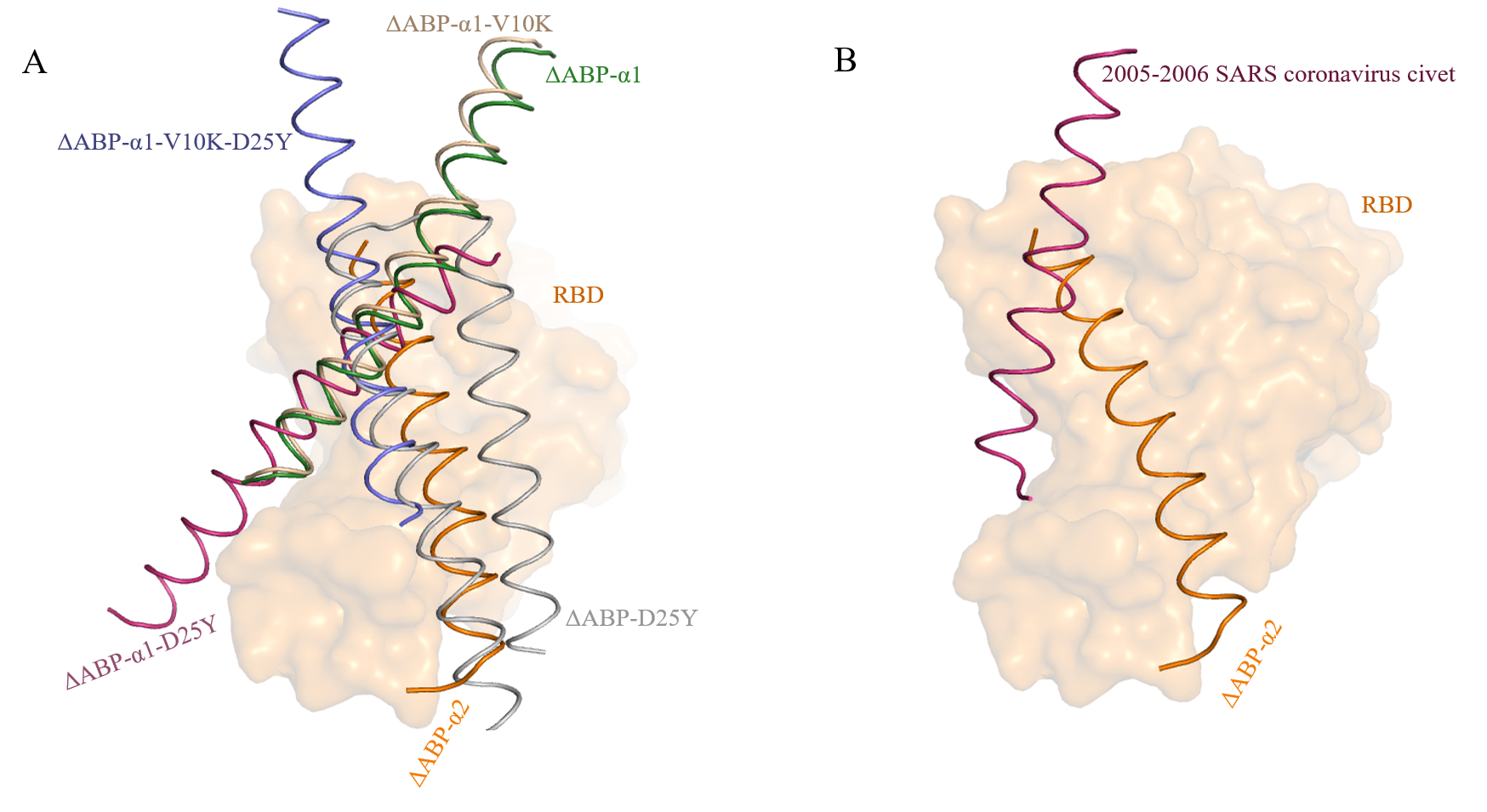
The previously identified inhibitor (ΔABP-D25Y) of RBD has two helices (α1 and α2) homologous to PD domain of hACE2 49. The inhibitor (ΔABP-D25Y) binds to RBD with higher affinity compared to hACE2. To further examine the role of individual helices of ΔABP in interaction with RBD, we examined following peptides - ΔABP-α1, ΔABP- α2, ΔABP- α1-V10K, ΔABP-α1-D25Y, and ΔABP-α1-V10K- D25Y. The mutations (D25Y and V10K) were introduced to complement the α1 helix of hACE249. We docked all peptides into SARS-CoV-2 RBD. The interaction restraints were generated using CPORT and BIPSPI servers 40,41. Both servers predicted peptides binding at RBM site of RBD. Therefore, RBM site residues (483-507) and all residues of peptides were used as active residues for docking. Best complex structures were selected based on cluster size and HADDOCK score. HADDOCK score is computed as a weighted sum of van de Waals, electrostatic, desolvation, and ambiguous interaction restraints energies. The ΔABP-α2 showed the best HADDOCK score (Table 1). ΔABP-α2 peptide sits between the two helices of ΔABP-D25Y (Fig 1A and Supplementary file 1). The N terminus and C-terminus of peptide are shifted about 8.9 Å and 3.2 Å respectively, towards RBD in comparison to α2 helix ofΔABP-D25Y. Thus, the ΔABP-α2 is closer to RBD compared to ΔABP-D25Y. The ΔABP-α2 peptide partially overlap with α1 helix of hACE2 (Fig 1B). The fragment up to residue Ile56 (His34 of hACE2) superimpose very well. From residue Glu35 (hACE2) onwards the α1/hACE2 deviates away from SARS-CoV-2 RBD. However, ΔABP-α2 peptide lies parallel to RBM throughout its length (Fig 1B). Hence the bound peptide showed higher binding affinity for SARS-CoV-2 RBD.
However, the ΔABP-α1 peptide and its mutants bind SARS-CoV-2 RBD with lower affinity (Table 1). Further, the ΔABP-α1 peptides bind at a different position than the α1 helix of ΔABP-D25Y (Supplementary Fig 1A). The ΔABP-α2 peptide also binds RBD of other coronaviruses at the same site. Binding score varies depending on the RBM site residues. The ΔABP- α2 peptide binds 2005-2006 SARS coronavirus civet strain RBD (pdb 3d0i) with best HADDOCK score (-85.6 +/- 0.2). The binding score is better than SARS-CoV-2 RBD. However, the bound peptide does not cover the whole RBM site (Supplementary Fig 1B). Thus, the proposed inhibitor has the potential to block the broader range of coronaviruses.
Binding affinity:
The binding affinity of the therapeutic agents to the receptor is an important parameter in predicting their effectiveness as promising drug candidates. To compute the binding affinity of peptides, we use PRODIGY server. The binding affinities of induvial peptides are listed in table 1. The predicted affinities are in accordance with HADDOCK score. The PRODIGY server predicted a dissociation constant (KD) of 2.6 nM (ΔG -11.7 kcal mol-1) and 0.049 nM (ΔG -14.1 kcal mol-1) for ΔABP- α1 and ΔABP- α2 peptides, respectively. The predicted KD of ΔABP-D25Y is 0.6 nM (ΔG -12.6 kcal mol-1) 49. The dissociation constants of mutant ΔABP- α1-V10K, ΔABP- α1-D25Y, and ΔABP- α1-V10K-D25Y are 16.0 nM (ΔG -10.6 kcal mol-1), 5.2 nM (ΔG -11.3 kcal mol-1), and 69 nM (ΔG -9.8 kcal mol-1) respectively. The order of peptides affinity to SARs-CoV-2 RBD is ΔABP- α2 > ΔABP- α1 > ΔABP-D25Y > ΔABP- α1-D25Y > ΔABP- α1-V10K > ΔABP- α1-V10K-D25Y. The experimentally determined KD for hACE2 to ectodomain S protein interaction is 4.7 nM 5,24. Thus, ΔABP- α2 peptide binds SARS-CoV-2 RBD with higher affinity than hACE2. A designed inhibitor should have a selective binding at the target site with relatively high binding energies. Thus, HADDOCK score and predicted KD suggests that ΔABP-α2 peptide binds RBD with remarkably high affinity.
ΔABP-α2 Complex:
The peptide ΔABP-α2 is parallelly aligned with RBM β sheet and covers the whole RBM site (Fig 1). The N-terminus of peptide interacts with the RBD capping loop (472-488) and C-terminus of peptide interacts with RBD loop (498-505). Thus, ΔABP-α2 peptide forms extensive contact with SARS-CoV-2 RBD. Sequence analysis of coronaviruses RBDs suggest that SARS-CoV-2 RBD has unique residues- Leu455, Phe486, Gln493, Ser494, and Asn501. These unique residues form extensive contact with hACE2 and responsible for higher affinity of SARS-CoV-2 RBD to hACE220,21,50. Thus, the designed inhibitor should disengage these residues from hACE2 interaction.
Leu455 of SARS-CoV-2 RBD enhances the viral binding to hACE2 because of its favourable interactions with hotspot 31 (Lys31 of hACE2)10. In SARS-CoV-2 RBD, Leu455 interacts with Asp30, Lys31, and His34 of hACE2. In RBD/ΔABP-α2 peptide complex, Leu455 interacts with GLU52, ASN53, Lys55, and ILE56 (Fig 2A). In SARS-CoV-2 RBD, Phe486 does not interact with α1/hACE2. Capping loops which harbours Phe486 is flexible and deviates towards bound peptide in ΔABP-α2 complex. In ΔABP-α2, Phe486 is coordinated by triad Leu43, Asn46, Asn47. The phenyl ring of Phe486 is sandwiched between side chain of Asn46 and Asn47 (Fig 2B). In SARS-CoV-2 RBD, Gln493 forms salt bridge interactions with Lys31 and Glu35 of hACE2. In ΔABP-α2/RBD complex also, Gln493 forms similar strong salt bridge interaction with ASN53 of peptide (Fig 2C). Ser494 does not involve in any interaction in SARS-CoV-2 RBD and hACE2 complex. However, in ΔABP-α2 complex Ser494 forms interaction with Val60 of ΔABP-α2 enhancing peptide interaction with viral RBD (Fig 2C). Lastly, Asn501 in SARS-CoV-2 RBD/hACE2 complex interacts with Tyr41 of ACE2. However, Asn501 interacts with Arg62 and Ile63 of peptide in. ΔABP-α2 (Fig 2D). Thus ΔABP-α2 can from the very strong interaction with RBD.
MD Simulation:
To understand the interactions between viral RBD and ΔABP-α2 100 ns classical MD simulation of the complex was performed. To assess the dynamic behaviour of the complex, the time dependent root-mean-square deviation (RMSD) of all protein atoms was calculated using original docked complex as reference. The RMSD value of the whole complex fluctuates between 0.3-0.4 nm suggesting flexible nature of the complex (Fig 3A). Both the RBM site and ΔABP-α2 peptide exhibit RMSD value of ~0.2 nm. Thus, the interface between peptide inhibitor and RBM is stable (Fig 3A).
The dynamic behaviour of protein at residue level was estimated by root-mean-square fluctuations (RMSF) calculation (Fig 3B). RMSF reflects the positions of the individual atoms with respect to the average position across the whole simulation trajectory. The peptide ΔABP-α2 residues are rigid and does not fluctuate much (RMSF < 0.15 Å) (Fig 3B). Similarly, the RBD residues are stable except some loops. Particularly two flexible regions are present in the RBD. The loop between (380-396) on the surface of RBD is flexible. This loop is far from the hACE2 interaction site. The second flexible region is the capping loop (476-490) near the hACE2 interaction site. The composition of this loop is remarkably different from SARS-COV. The following mutations Pro469/Val483, Pro470/Glu484, Thr468/Gly482, Cys467/Cys480, Lys465/Thr478, Asp463/Gly476, Pro462/Ala475 (SARS-CoV/SARS-CoV-2) makes SARS-CoV-2 loop more flexible. An extra amino acid Asn481 in SARS-CoV-2 loop is present. The absence of two Pro residues, presence of Gly and Cys, and insertion of Asn481 in SARS-CoV-2 make the capping loop more flexible (RMSF ~ 0.8 nm) (Fig 3B). However, interface residues showed overall low RMSF (0.1-0.2 nm) and all critical amino acids maintain their interactions with ΔABP-α2 peptide. The single α helical peptide inhibitor of SARS-CoV-2 based on PD domain of hACE2 was found to be less stable51. However, ΔABP-α2 peptide retain its shape and provide full coverage to the RBM site (Fig 3B).
Radius of gyration (Rg) is the measure the compactness of the molecule in the solution. The Rg SARS-CoV-2 RBD and ΔABP-α2 peptide complex fluctuates between 1.87-1.97 nm. Significant variation is seen at around 50 ns simulation which can be attributed to the flexible loops of RBD (Fig 3C). Similarly, solvent accessible surface area (SASA) curve supports the Rg plot (Fig 3D). Thus, classical MD simulation studies suggest that the interface of the complex is stable, and the inhibitor ΔABP-α2 does not dissociate from RBD. A good peptide inhibitor should have selective binding to the receptor, complementary shape, and low flexibility of the interface residues 52.
Principle component analysis (PCA):
PCA is a standard method to characterize the variable correlations from atomic fluctuations in an MD trajectory. PCA can provide a brief description of the motions. PCA extracts highly correlated motions from MD trajectories using dimensional reductions. To understand the motion of ΔABP-α2 peptide, RBM site, and capping loop, PCA analysis was performed (Fig 4). The core of the RBD, the bound peptide, and RBM site do not show any motion (Fig 4A). The C terminus of peptide showed slight twisting motion and shifted away from RBD significantly (~ 6.0 Å). These residues are not involved in direct interaction with RBD. Without complete protein fold, the isolated helical peptides are usually unstable which in turn reduces the affinity of the peptides to the target protein53. The peptide ΔABP-α2 maintained its helicity and remained bound to viral RBD throughout the simulations. The pose of peptide did not change during the simulation. Specially, the central portion of peptide ΔABP-α2 maintains its interaction with viral RBD (Supplementary file 2).
However, the capping loop (476-490) of the peptide showed highest degree of motion (Fig 4B). The fluctuates between open and close conformations (Fig 4B and Supplementary File 2). The ΔABP-α2 peptide maintains its interaction with capping throughout its transition from open to closed state. The second significant motion occurs in the fragment between residues (358-394) of RBD. This region is on the surface of RBD and far from the interface of RDM and ΔABP-α2 peptide (Fig 4C).
Immunoinformatics:
The peptide inhibitor is recognised as a foreign substance by the human host cells, thus inducing a host immune response54. The antibodies can affect the efficacy of the drug by reducing the lifetime, neutralising the activity, and altering the pharmacokinetics of the drug. Thus, an ideal inhibitor should not be antigenic. The ΔABP-α2 peptide was not antigenic according to VaxiJen 2.0 server 46. The server predicted protective antigen score of -0.0046, which is much lower than the threshold 0.4. The B cell epitope is a portion of an antigen recognized by either a particular B cell receptor or the elicited antibody 55. There are two types of B cell epitopes -1) Continuous and linear and 2) Discontinuous or structural. More than 90% of B cell epitopes are structural 56–58. DiscoTope analysis suggests the propensity scores of the individual residues for discontinuous B cell epitope (Fig 5A).
The recognition of viral peptides-MHC class I complex by CD8+ T cells is necessary for antiviral immunity. The T cell epitopes are necessary to design an effective vaccine, however, will pose a challenge for peptide-based drugs. We used NetCTL 1.2 server to predict Cytotoxic T lymphocyte (CTL) epitopes (Fig 5B). This tool outcomes combined score of MHC class I binding, proteasomal C terminal cleavage and TAP transport efficiency. The highest score (0.5391) was seen for peptide (LVENNRLNV). Thus, no CTL epitopes were predicted on ΔABP-α2 sequence. Thus, above analysis suggest the ΔABP-α2 is non immunogenic and will be a probable candidate for therapeutic use.
{kind=link}