The bacterial strain
Citrobacter freundii EGY-NE1 was isolated from Egyptian clay soil (Damietta) and identified according to its phenotypic and molecular characters (Frederiksen 2005) by Rizk (2019).
Growth medium and conditions
Citrobacter freundii EGY-NE1 was cultured on tryptone-glucose-yeast extract broth medium (tryptone 5.0 g, yeast extract 5.0 g, asparagine 10 g, glucose 1.0 g, K2HPO4 1.0 g; pH 6.0) at 37 °C and 75 rpm for 48 hours. The supernatants of grown cultures were separated by centrifugation at 7,000 rpm for 10 minutes.
L-asparaginase assay
We determined the L-asparaginase activity by assaying the released ammonia during the hydrolysis reaction of asparagine by the enzyme extracts (Prakasham et al. 2009). A mixture of the enzyme extract (0.1 ml), Tris-HCl buffer (0.2 ml, 0.05M, pH 8.6) and L-asparagine solution (1.7 ml, 0.01M) was incubated for 10 minutes at 37°C. The reaction was stopped by adding 0.5 ml of 1.5 M trichloroacetic acid followed by centrifugation at 1000 rpm; 0.5 ml of the supernatant was diluted to 7 ml with distilled water and 1 ml of Nessler’s reagent was added. After 10 minutes, the absorbance of the developed color was measured at 480 nm by a spectrophotometer (model UV1100, Shanghai Yoke Instrument Company, China). One unit of L-asparaginase was defined as the amount of enzyme that liberates one µmole of ammonia per minute (Prakasham et al. 2009).
Protein Estimation
Total protein was estimated according to the method of Bradford (1976).
Purification of L-asparaginase from Citrobacter freundii
Proteins in the crude enzyme preparations were precipitated under cold conditions by using different concentrations of ammonium sulfate (40, 50, 60, 70, and 80%) with stirring for 20 minutes; then, we left it overnight at 4°C for complete precipitation. The precipitated proteins were separated at 5000 rpm for 20 minutes then suspended in sodium phosphate buffer (0.1M, pH 7). These enzyme preparations were put in dialysis tubes against 0.05 M sodium phosphate buffer (pH 7) overnight at 4˚C to get rid of salts.
Gel filtration chromatography followed by anion exchange chromatography was used to purify the enzyme. The enzyme preparation was applied to a gel filtration Sephadex G-50 column (55 × 1.5 cm) equilibrated by Tris-HCl buffer (0.05 M, pH 8), then eluted by the same buffer. The eluted fractions (2 ml) were assayed for L-asparaginase activity and protein content. Active fractions were pooled for further purification by anion exchange chromatography.
The active fractions were applied to an anion exchange column of DEAE-cellulose (30 × 1.5 cm) equilibrated with Tris-HCl buffer (0.05 M, pH 8). The proteins were eluted with a linear gradient of 0.0 to 1.0 M NaCl in the same buffer; each fraction (2 ml) was assayed for L-asparaginase activity and protein content.
Sodium dodecyl sulfate poly-acrylamide gel electrophoresis
Sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE) was used for qualifying and comparing protein samples according to the method of Laemmli (1970). Protein samples were boiled for 2 minutes in the sample-loading buffer. The molecular weight of protein bands was referenced to a pre-stained protein marker (# SM 0671, Fermentas). Electrophoresis was carried out by Bio-Rad Mini-Protein II cell gel apparatus at 120 volts.
The separated proteins on the gel were stained in Coomassie brilliant blue R-250 for one hour; then, soaked in a de-staining solution (H2O: methanol: acetic acid 50:40:10) for two hours until the protein bands appeared.
Activity staining of L-asparaginase
Activity staining was performed in a Petri dish containing L-asparagine gel (1% L-asparagine, 1% agar dissolved in 25 ml of 0.05 M Tris-HCl buffer; pH 8.0). We added the enzyme solution (20 μl) to a formed well in the gel; then, incubated it at 37°C. After 30 minutes, the gel was stained with Nessler’s reagent; the formation of a brown zone indicates L-asparaginase activity (Lincoln et al. 2019).
Biochemical and kinetic properties of the purified L-asparaginase
We studied the enzyme activity at various temperatures by incubating the assay reaction mixtures at different temperatures (31, 34, 37, 40, 43 and 46 °C). To study the effect of pH on L-asparaginase activity, we adjusted pH of Tris-HCl buffer (the standard buffer of the assay) to different pH values (3, 4, 5, 6, 7, 8, 9 and 10). The activities were expressed as a relative percent to the maximal activity.
The effects of metal salts (NaCl, KCl, CaCl2, ZnCl2, BaCl2, MgCl2 and CaSO4) and inhibitors (sodium azide, sodium tartrate, mercuric chloride, EDTA and sodium EDTA) on enzymatic activity were investigated. The purified enzyme (0.2 ml) was mixed with the salt or inhibitor solutions at 10 mM final concentration; then, incubated at room temperature for 30 minutes before the enzyme assay. The activities were expressed as a relative percent to the control.
For the kinetic properties, the enzyme activity was assayed by using reaction mixtures containing different L-asparagine concentrations (9, 10, 20, 30, 40, 50 and 60 mM). The Km and Vmax values were calculated from Lineweaver–Burk plots.
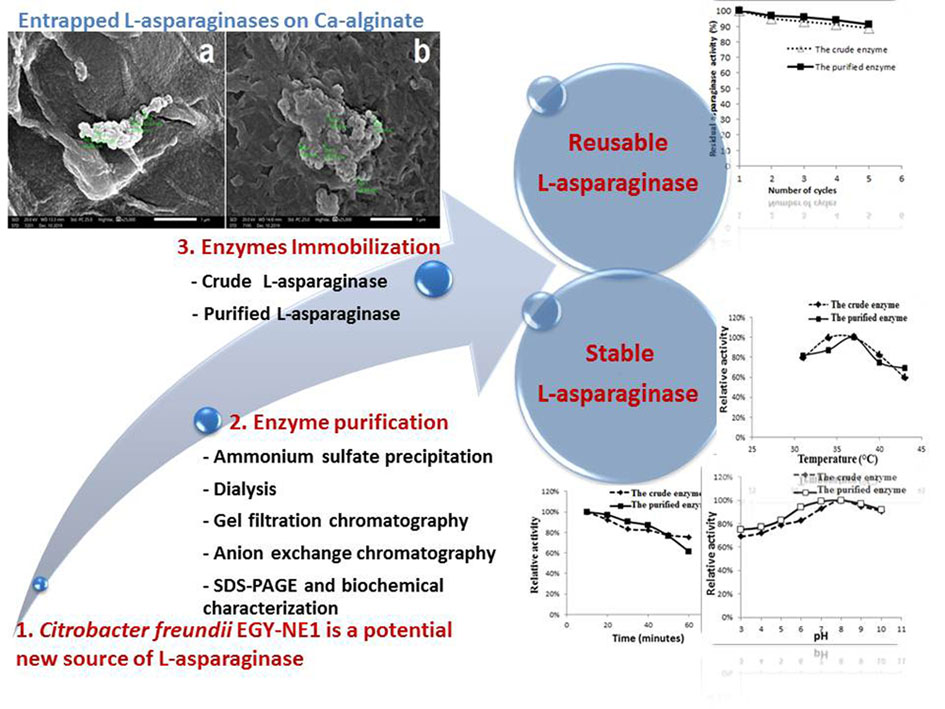
Immobilization of the crude and purified L-asparaginase by encapsulation in Ca-alginate
The enzyme was encapsulated in Ca-alginate beads as described by Ulu and Ates (2017). We mixed the crude or purified L-asparaginase (10 ml) with Na-alginate solution (40 ml, 2%); this mixture was extruded (from 3 cm controlled distance) in 2% CaCl2 solution using a micro-pipette. After 30 minutes, the formed beads were filtrated; then, washed with a sterile NaCl solution (0.9%) followed by sterile distilled water (three times). The protein content and enzymes activity were assayed after immobilization.
Characterization of the immobilized enzymes morphology
Morphology of immobilized crude and purified L-asparaginase was examined by scanning electron microscope (SEM, JEOL, JSM IT200, Japan).
Effect of temperature and pH on the immobilized crude and purified L-asparaginase
To determine the effect of temperature on the immobilized crude and purified L-asparaginase, the assay reaction mixture was incubated at different temperatures (31, 34, 37, 40, 43 and 46 °C). The effect of different pH was also studied by adjusting pH of the standard assay buffer to different values (3, 4, 5, 6, 7, 8, 9 and 10). The thermal stability of the immobilized crude and purified L-asparaginase was studied at 40˚C. The enzyme beads were incubated at 40˚C for 60 miutes, without a substrate; the enzyme activity was assayed at 10 minutes intervals. The activities of the enzyme were expressed as a percent of the maximal activity.
Reusability of the immobilized enzyme
The reusability of the immobilized crude and purified enzyme were tested up to 5 rounds; the enzyme beads were incubated with the reaction mixture at optimum conditions for the assay; at the end of the reaction, the beads were removed from the reaction mixture and washed with distilled water; these beads were repeatedly used in the subsequent four assay rounds. The activities of the enzyme were expressed as a percent of its initial activity.
{kind=link}