Study setting and participants {9}
The study is conducted in nine renal dialysis units across the NT. First Nation Australians on MHD for over 3 months are recruited in the study. Participating units include the 7A Dialysis Unit at the Royal Darwin Hospital, Nightcliff and Palmerston renal dialysis units in Darwin, Katherine Dialysis unit, Tiwi Islands Dialysis unit, Tennant Creek Dialysis unit, the inpatient renal ward at Alice Springs Hospital and the Gap Road and Flynn Drive Dialysis units in Alice Springs.
Eligibility criteria {10}
Participant inclusion criteria include: 1) Male or female aged ≥18 years old 2) Identify as Aboriginal and/or Torres Strait Islander (First Nation) 3) On maintenance haemodialysis for ≥3 months 4) the following clinical laboratory results: a) haemoglobin ≤115g/L, c) ferritin levels between ≥700µg/L & ≤ 2000µg/L d)TSAT <40% e) CRP <50mg/L 5) Willing to join the study and can provide informed consent and 6) on adequate erythropoietin stimulating agent (ESA) therapy
The following are the participant exclusion criteria: 1) History of known allergic or adverse or hypersensitivity reactions to iron polymaltose. 2) Already receiving iron unless stopped for ≥4 weeks at the time of recruitment 3) Have received blood transfusion within the last 4 weeks 4) Known iron overload, haemochromatosis, haemoglobinopathy, haemolytic anaemia, aplastic anaemia, lymphoproliferative disease or cancer or on current cancer treatment 5) Participant’s primary clinician unwilling to enrol 6) Not planning to remain resident in the NT for at least 12 months 7) Ferritin >2000𝜇g/L 8) TSAT ≥40% 9) CRP ≥50mg/L 10) Life expectancy <6 months based on the judgement of the investigator 11) Living-donor transplant scheduled within 12 months 12) Scheduled to switch to peritoneal dialysis
Temporary Exclusion Criteria: Participant may not enter the trial at any given time if fever >380C, or has evidence of active bacterial infection, or CRP ≥50mg/L and current severe acute asthma, eczema, and any allergies.
Interventions {11}
Treatment of Study Participants: Participants are randomised to either Arm A: Intervention IV Iron treatment group (Figures 1 & 2). or Arm B: Control group receiving no intravenous iron treatment (standard treatment) (Figures 1 & 2). After the randomisation, participants receive monthly treatment as per their randomisation of either monthly intravenous iron if ferritin ≥700µg/L and ≤2000µg/L and TSAT <40% (Arm A) or no IV iron treatment (Arm B) for up to 42 months. Study treatment is withheld if monthly testing demonstrated ferritin >2000μg/L and/or TSAT ≥40%. These markers will be measured prior to administration of Arm’s A or B treatment. For both arms A and B, if the ferritin level is <700µg/L and TSAT <40% rescue therapy is administered. All treatment is administered during dialysis via the ports on the dialysis circuits. (See additional file 2)
Intervention treatment Arm A and the control Arm B {11a}: Intravenous iron polymaltose is administered according to the standard clinical protocol during haemodialysis. For arm A, patients receive IV iron polymaltose 200 mg during each of the first 2 haemodialysis sessions at the start of the study, and during each of the first 2 dialysis sessions of the week following the monthly blood tests for all subsequent months (i.e., 400 mg per month). If monthly testing demonstrates ferritin > 2000μg/L and/or TSAT ≥40%, IV iron is withheld for the month. The IV Iron is restarted as per protocol when the monthly tests demonstrate Ferritin >700µg/L & ≤2000µg/L and TSAT <40%. Rescue therapy of 400mg of Iron Polymaltose is administered intravenously on haemodialysis if ferritin ≤700µg/L and TSAT <40% or as indicated by the treating nephrologist (See below). If monthly testing demonstrates ferritin >700μg/L &≤2000 and TSAT<40%, patients will continue with their allocated randomised treatment (Figure 2).
Criteria for modifying allocated interventions {11b}: Rescue therapy: For both arms A and B, if monthly blood tests demonstrate ferritin <700µg/L and TSAT <40%, 200 mg of intravenous iron polymaltose is administered during each of the first two haemodialysis sessions in the second week after the monthly blood tests. When the monthly blood tests indicate ferritin >700µg/L & ≤2000µg/L and TSAT <40%, the rescue therapy will be ceased, and participants continue to receive their allocated study treatment.
Withholding of therapy: For both arms, if monthly blood tests demonstrate ferritin >2000μg/L and/or TSAT ≥40%, study treatment is withheld. When the monthly blood tests indicate ferritin ≤2000µg per litre and TSAT <40%, patients continue to receive their allocated study treatment.
Strategies to improve adherence to protocol {11c}: Training of site nurses and nephrologists: All site staff at each participating dialysis unit are trained in the study protocol, Standard operating procedures (SOPs) and their reporting requirements by the Clinical Trial Manager or trained research nurse, a study chief investigator or delegate, prior to the site being opened for recruitment. All site investigators complete a computer-based training course in Good Clinical Practice and assist in providing robust training to their respective site staff.
The Clinical Trial Manager or delegate have regular phone, email or video conferencing contact with each participating dialysis unit.
Relevant concomitant care that are permitted or prohibited during the trial {11d}: ESA therapy: All patients should be on adequate ESA dose at the time of randomisation. The ESA dose is determined according to the routine ESA dosing protocol. ESA dosing can be adjusted by the caring clinical teams as per the usual ESA administration protocol.
Documentation in patient’s medical record: A sticker is placed in the patient’s dialysis medical record (one on the progress notes on the day of randomisation, and one in the front inside cover of the dialysis record and on the medical record [“old notes”] if one exists). This sticker will alert clinicians that the patient has been randomised to the INFERR study, with a brief explanation of the study, and confirmation that the participant has provided written informed consent.
A copy of the study synopsis is also placed in the medical notes of the patient. A checklist of study procedures is also placed in the notes.
For electronic medical records, an electronic “sticker” is used, and appropriate annotations are made in the Primary Care Information System (PCIS) as well as letters in the Clinical Workstation (CWS). This is routine practice for all patients within the NT renal services.
Checking of medical records: The medical records (paper or electronic) are checked 3 months post randomisation (+/- 1 month) and at least 3 monthly (+/- 2 months) thereafter for up to 36 months by the study team to ensure adherence to the study protocol.
Study Outcomes {12}
All outcomes will be measured from the time of randomization to the end of the follow-up period.
The primary outcome: the differences between the two-study arms in the risk of hospitalisation with all-cause infection or death.
Secondary outcomes:
- Differences in Hb level at three, six, twelve and 24 months and over the follow-up
- The differences between the two-study arms in: A composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for heart failure or death from any cause
- All-cause death
- Hospitalisation with any new diagnosis of infections
- Hospitalisation with a principal diagnosis of infection
- Vascular access thrombosis
- Peripheral vascular disease and/or amputations
- All-cause hospitalization
- Fatal and nonfatal cardiovascular event (MI, stroke, and hospitalization for HF)
- Fatal or nonfatal MI
- Fatal or nonfatal stroke
- Heart failure hospitalization
- Length of hospital stay
- ESA resistive Index (ERI), defined as average weekly erythropoietin (EPO) dose per kg body weight (wt) per average haemoglobin (Hgb), over a 3-month period (ERI = (EPO/wt)/Hgb))
- Serious adverse events
- all serious adverse events
- all hospital admissions
- infections associated with hospitalization.
- all-cause mortality
- Iron overload (measured by MRI FerriScan@ of the liver)
- Cost analysis: an analysis of the financial costs of the treatment will be performed assessing the costs of ESA, iron, hospital admissions and procedures for the duration of follow-up among all patients assigned to intravenous iron compared to no iron treatment.
Outcomes assessed with non-randomized analyses (i.e., using INFERR as a longitudinal cohort study)
1) Factors associated with high serum ferritin levels: Association between ferritin and: a) other measures of iron status (serum hepcidin, TSAT, soluble transferrin receptor, percentage hypochromic red cells, reticulocyte haemoglobin content, liver iron), b) Inflammatory markers (CRP, Tumour Necrosis Factor alpha, IL1 and IL6) and c) others (infection rates, CV events, hepatitis, medications, comorbidities, dialysis vintage, dialysis access, hospitalisations, time variation of ferritin)
2) Assessment of liver disease: LFTs, Hepatitis B status, ultrasound and fibroscan of the liver
3) Assessment of the comparability of the current ferritin assay platforms: the Abbott ARCHITECT versus the Vitros® Ortho Clinical Diagnostics (OCD) platforms
4) Analysis of defined primary and secondary outcomes: by average weekly dialysis attendance (according to both ordinal attendance group and using attendance as a continuous variable)
5) Analysis of defined primary and secondary outcomes: by markers of inflammation
Tertiary outcomes comparison between the two treatment groups.
- Cumulative dose of iron
- Serum Ferritin levels
- TSAT
- Platelet count
- Serum albumin levels
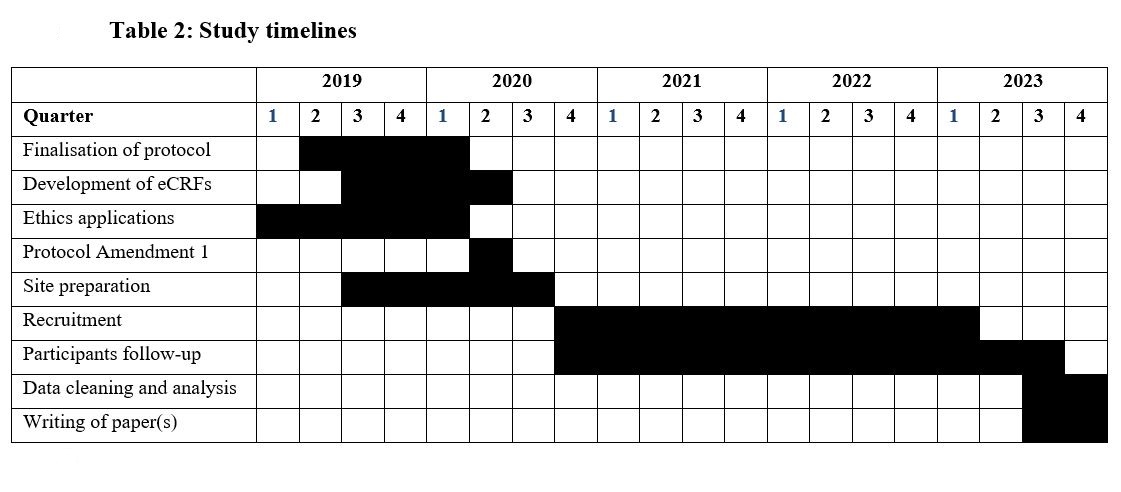
Participant timeline {13}
See figure 2 and table 1 & 2.
Sample size {14}
Maximal planned follow-up in the study will be 36 months (3 years). The 2-year rate of admission with first episode of infection or death in our patients on maintenance haemodialysis from 2000-2014 was 60%. In order to achieve a study power >80% at 5% significance level in detecting a 30% reduction (from 60% to 48%) in the rate of first hospitalisation with all-cause infection or death, using the log-rank test (time to event modelling) assuming a 5% drop-out rate, 576 participants will be randomised 1:1 into each arm. The study also has > 90% power at 5% significance level of detecting a difference in haemoglobin of 5g/L and standard deviation of 11.3g/L based on published data (44, 45). The sample size also provides enough power to determine differences within the ferritin strata.
Recruitment {15}
Screening: Patients receiving haemodialysis in renal units across the Northern Territory undergo a series of clinical laboratory tests (haematology and serum chemistry) at the commencement of the month as part of routine care. These results are used to screen potential participants for eligibility into the study.
The treating renal team are approached for a discussion on potential recruitment of the patients into the trial, and for permission to review the patient’s medical notes. The study team assesses the eligibility criteria and completes the Pre-Screening Case Report Form (CRF). If ineligible, the patient is not approached. If considered eligible the patient is approached for an informed consent discussion using the teach back method to determine if an interpreter is required to obtain consent. After consent is obtained, eligibility will be re-established prior to randomisation. Each unit maintains a log of all pre-screened patients along with their eligibility and consent outcomes in the medical records or notes. However, should a potential participant consent meet the temporary exclusion criteria at time of screening they will be re-approached for inclusion into the trial at a subsequent clinic visit.
To maximise recruitment, the research team spend a considerable time in the renal units providing information and education to potential participants using the teach back method. There is also a potential for expanding the recruitment to other renal units across Northern Australia to achieve the desired sample size.
Methods: Assignments of interventions (Randomisation and blinding)
Allocation
Sequence generation {16a}: Prior to proceeding with randomisation, the research nurse and/or investigator ensures informed consent has been obtained and documented and confirm that the participant is eligible to be enrolled. Participants are randomised using a module in the web-based study database, the Research Electronic Data Capture (Redcap) that is password protected and stored on a secure drive at Menzies School of Health Research. Compulsory fields required prior to randomisation are screening number (Study Identification number), confirmation of eligibility, age, hospital registration number (HRN), confirmation of consent, recruitment site (unit) and ferritin strata. Randomisation will be stratified by location (Top End and Central Australia) and ferritin strata (≤1200µg/L and > 1200µg/L) and will be in permuted blocks of variable block size. Participants are randomised in a 1:1 ratio to either Arm A: Intervention treatment group or Arm B: control group via Redcap, available 24 hours per day, 7 days per week (Menzies School of Heath Research, Darwin).
Allocation concealment mechanism {16b}: The randomised sequence allocation is stored on the password protected secure server by the trial statistician and will not be available to any investigators or member of study staff.
Implementation {16c}: Randomisation is performed at the site with allocation occurring via the web-based database (Redcap), this only occurs after informed consent is obtained and eligibility confirmed. Participants are enrolled by Investigators or research nurses that have been delegated this responsibility by the Chief Investigator (CI). To randomise the participant, the research nurse or investigator logs onto Redcap and enters the details required to obtain the treatment randomisation number assigned to the participant for that site.
Blinding (masking) {17a}: This is an open-label blinded end point assessment trial. The intravenous iron treatments are supplied by a central pharmacy, either at the Royal Darwin Hospital or the Alice Springs Hospital and is prepared and administered by the nursing staff as per routine clinical care. The patient, nurses administering the drugs and investigators will not be blinded to the treatment allocation. The administration will be direct into the dialysis circuit as per the standard intravenous iron administration procedure on haemodialysis.
The investigators assessing and analysing the primary outcomes (see below –outcomes assessment committee) are blinded to treatment allocation. Independent outcomes Assessors not involved in any aspect of the study will review and complete a data collection form of all randomised study participants to independently determine the study outcomes (from both electronic and written medical records). In view of the relationships likely to develop between the research staff and some of the participants, it is essential that we engage independent outcomes assessors. The outcome assessment data collection forms and final assessment will be verified by two of the Chief Investigators.
Procedures for unblinding outcomes if needed {17b}: An Independent Data Safety and Monitoring Board (DSMB) reviews safety data, including serious adverse events and the primary endpoint events, and will advise on continuation of the study, or whether the study should be stopped prematurely because of safety concerns. In a closed session, the DSMB will be provided with descriptive analyses of study data by study labels “A” and “B” for the allocated treatment arms.
Methods: Data collection, management, and analysis
Data collection methods and data management {18 & 19}
Assessment and collection of outcome, baseline, and other trial data {18a}: Source documents are those where data are first recorded, and from which participants’ CRF data are obtained. These include but are not limited to hospital records both electronic and paper (which will include medical history, previous and current medications, any relevant radiography test, blood test results, haemodynamic parameters, and medical correspondence) and paper or electronic clinic records (which include vital status, recent medical history & relevant blood culture results). A further potential data source will be through telephone conversations with the study participant or General Practitioner.
Storage and archiving of study documents (CRFs and consent forms) are the responsibility of the site principal investigator and these will remain at the site of recruitment. All study participants are allocated a unique number at time of screening (screening number or study ID number), this screening number is added to all the CRFs for that participant. The participants also have their HRN recorded on the CRFs as this information is required to ensure the correct medical record is accessed during medical record reviews.
Data for this study is recorded via a secure, Electronic Data Capture (EDC) web-based system (Redcap). It is transcribed by the study team from the paper CRFs into Redcap. Data is stored in a re-identifiable manner in the database, using the unique screening number for each patient. The database contains validation ranges for each variable to minimise the chance of data entry errors. An audit trail will maintain a record of initial entries and changes made; reasons for change; time and date of entry; and username of person who made the change. Data queries are raised by the Clinical Trial Manager and study monitor, and missing data or suspected errors are raised as data queries and resolved prior to database lock and analysis. The database contains in-line capability so that these queries and answers are logged as part of the audit trail.
For each potential participant screened (including screen failures), the screening eCRF is completed by the study team. For each participant enrolled, eCRFs must be completed. This also applies to records for those patients who fail to complete the study. The study team should ensure the accuracy, completeness and timeliness of the data reported in the eCRFs and in all required reports. A comprehensive validation check program verifies the data and automatically generates discrepancies for resolution by the study team. Manual discrepancies can also be raised if necessary.
In addition, for selected data fields, accurate and reliable data collection is assured by verification of the eCRFs against the study team’s records by the study monitor (source document verification), and the maintenance of medication compliance is captured in the CRFs from the participant’s medication chart (source document) by the study team.
Blood samples and data collection
Data Collection from routine care: Routine blood tests: Haemodialysis patients have monthly blood tests as part of their routine care. Tests obtained as part of this routine screen that are analysed as outcomes and covariates of interest for this study include: Hb, CRP, full blood count (will include MCV and MCHC), liver function tests, iron studies (ferritin and TSAT), measures of dialysis adequacy (Urea reduction ratio and KT/V (a number calculated from change in urea pre- and post-dialysis, time on dialysis and volume of distribution of urea used to quantify dialysis adequacy). Every 3 months the following are also routinely collected: parathyroid hormone levels and glycated haemoglobin (HbA1c).
Additional blood tests: Samples are collected at baseline and at 12 months for: 1) other measures of iron status (serum hepcidin levels, soluble transferrin factor levels, percentage hypochromic red cells, reticulocyte haemoglobin content); 2) other causes of anaemia (thyroid function tests, vitamin B12 and folate (only at baseline); 3) Inflammatory markers (serum Tumour Necrosis Factor (TNF) alpha, Interleukin-1 (IL-1) and Interleukin-6 (IL-6); and 4) and marker of the metabolic syndrome (serum total adiponectin).
Assessment of the comparability of the two ferritin concentrations measurements methods: From samples collected at around 1 month comparison of the Abbott ARCHITECT i1000 (Abbot Diagnostics, USA) versus the Vitros Ortho® XT7600 in a sample of about 150 participants covering the analytical range and above of serum ferritin levels. Ferritin concentrations in the NT were werev previously been determined using the Abbott ARCHITECT which is the platform used by most laboratories in Australia. Territory Laboratories have recently changed to and now routinely use the Vitros Ortho Clinical Diagnostics platform for analyses of all biochemical analytes. The Vitros Ortho XT7600 gives ferritin concentrations results up to 40% lower than the Abbott ARCHITECT platform. Determination of iron overload: In addition to serum ferritin and TSAT levels, liver iron levels will be determined by use of the non-invasive but accurate method; the magnetic resonance imaging (MRI) based technique spin-density projection-assisted (SDPA) R2- MRI (FerriScan®) at baseline and at 12 months.
Demographic and clinical data: A full and relevant clinical and social history will be obtained from each participant. Key factors of direct interest for this study include time on dialysis, dialysis access and dialysis access interventions, ideal dry weight, height, body mass index, waist hip ratio, primary renal diagnosis, comorbidities (liver, lung, diabetes, cardiovascular etc.), current smoking status, history of current or previous alcohol intake, dietary history, any significant illnesses, previous Hb levels, a full medication list and history, and sociodemographic data.
Other data from routine care: all patients are routinely followed up by their nephrologist every 3 months and data from these follow up appointments is reviewed.
Additional follow-up for the study: all participants have medical reviews by the study team at 3, 6, 12, 18, 24, and 36 months (primary outcome assessment).
Major events: Hospitalisation due to all-cause infections or death are documented as the primary outcome. All admissions overnight or longer are followed up with collection of detailed data on the admission. Data on all major vascular events, all dialysis access-related events including infections and vascular interventions not requiring hospital admission are also collected.
Laboratory procedures: All blood tests are processed as per the local laboratory’s usual procedures. Some samples will need to be processed by the Menzies laboratories and Western Diagnostics as per standard clinical practice. Please refer to the INFERR Laboratory Standard Operating Procedure (SOP) for further information (Additional file 3).
Storage and testing of biological specimens: To ensure availability of relevant samples for study procedures, the laboratory linked to each site will freeze and store some samples. These will later be transported for archiving in -80 degrees centigrade freezers at the Menzies School of Health Research laboratory and Territory Pathology laboratories at Darwin and Alice Springs. Samples are transported to Menzies in batches at four specified time points in the study – 3 months after the recruitment of participant # 100, 200, 300, 400 and 576. Samples are identified by their INFERR study number and local laboratory specimen ID number.
Plans to promote participant retention and follow up {18b}: The study team makes regular contact (either by phone or preferably in person on the units) with the participants’ treating teams at 3 months and every 6 months. The purpose of this contact is to check compliance with the protocol in terms of study drug prescribing and ordering of routine clinical blood tests. Data is collected and entered into the corresponding visit CRF. Standard Operating Procedures (SOPs) will contain step by step details on how to recruit patients, collect data and monitor progress.
The composite primary endpoint is assessed by an independent blinded endpoint adjudication committee. This committee consists of three nephrologists appointed by the trial management committee. This committee is provided with an extract of study data that does not contain patient identifiers, and does not contain any mention of treatment allocation or any detail about treatment of any kind, but does contain data relating to adverse events, procedures and concomitant medication changes linked to the primary and secondary outcomes of the study:
1) Demographic details (such as age and gender)
2) Comorbidities
3) Clinical details
4) Date and result of all blood results
5) Date and result of all other available clinical information
6) Hospitalisations and/or deaths
The members of the committee may request more information if needed, but this will only be provided if it is available and does not provide direct or indirect evidence of treatment allocation. Each of the three members of the committee will then independently determine if, in their view, the patient has met the primary endpoint. If there is a discrepancy between the three assessments, the majority will determine the endpoint.
Participants have the right to choose to withdraw from the study at any time and the investigator may discontinue a participant from the study or from treatment if deemed appropriate at any time. This will have no impact on their normal dialysis and other treatment or care. Reasons why a participant may be withdrawn from the study include, but are not limited to, participant request, primary treating clinician’s request, participant was enrolled and is ineligible (either arising during the study or was overlooked at time of screening and enrolment). Participants will not be withdrawn due to adverse events. The decision to withdraw a participant from the study must be discussed with the coordinating investigators.
If the participant withdraws consent from participating in the study and also withdraws consent for collection of future information, no further evaluations will be performed, and no additional data will be collected. The sponsors may retain and continue to use any data or samples collected before such withdrawal of consent. Participants that are lost to follow up will continue to be followed by collecting as much data as possible, if possible, until the end of the trial to avoid missing data. Participants withdrawn from the treatment by the treating clinicians will continue to be followed up to the end of the trial to avoid missing data and will be used in the intention-to-treat analysis. This study has allowed for 5% of participant drop out. Withdrawn participants will not be replaced.
If a participant is withdrawn the reason is recorded in Redcap.
Statistical methods {20a}: Statistical analysis plan: Data will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines for reporting of randomised trials. Demographic, clinical, biochemical and outcome data will be described by allocation arm. Continuous normally distributed data will be summarised with mean and standard deviations (SD), continuous not normally distributed variables with median and inter-quartile ranges (IQR). Categorical variables will be described with frequency and corresponding percentage.
Methods for analysing primary and secondary outcomes {20a}: Primary and secondary endpoints will be analysed according to the intention to treat principles (All participants data will be analysed according to the treatment allocation, regardless of what treatment they received and either they completed the follow up or not).
The dichotomous primary outcome and all the dichotomous secondary events will be analysed using time to first event regression using Cox proportional hazard models where the duration of the follow-up will be censored at the first between: date of outcome of interest, end of study, date of withdrawal (including withdrawal due to change in renal replacement therapy modality) or lost to follow-up. Given that these are competing events, sensitivity analysis will include analysis using Fine and Gray regression models (49). The effect of the intervention will be estimated as compared to placebo using Hazard Ratios with corresponding 95% confidence intervals and p-values. Recurrent event outcomes (i.e., rates of hospitalization) will be analysed using the method of Ghosh and Lin (50) or, as an alternative, using random effects log-binomial models or one of the extensions of the Cox proportional hazards model (51, 52). The approach choice will be guided by the distribution of the time to event and results of goodness of fit statistics.
Continuous outcomes assessed at multiple time points (e.g., haemoglobin) will be analysed using linear mixed models with a fixed effect for study allocation arm and time point and a random intercept to model the correlation of measurement taken within the same individual.
All the models will be univariable and include treatment arm only as independent variable (linear mixed models will also include a variable for time each outcome repeat was recorded).
Sensitivity analyses using a multivariable model to adjust for potential confounders will be performed when these factors appear to be not equally distributed between the two allocation arms.
Missing data will not be imputed. However, those with missing data will be described. This number is expected to be small (<5% of randomised patients) and the same in each arm as maintenance haemodialysis patients attend haemodialysis three times per week, almost no First Nation dialysis patients leave the Northern Territory, and we will follow patients who move back to their community.
Pre-defined secondary stratified analyses will be performed, comparing the effect of treatment between allocation arms according to:
i) The presence of active Hepatitis B carriage (defined dichotomously by the presence of either measurable Hepatitis B DNA according to PCR by the ABBOTT REALTIME HBV assay, or Hep B surface antigen positivity measured according to Immunometric Immunoassay (Serum), at any time during the study).
ii) Average weekly haemodialysis attendance over the time included in the study, defined as an ordinal variable [in three groups: <2.0 sessions/week, 2.0-2.75 sessions/week, >2.75 sessions/week] for stratified analysis, as well as incorporated in Cox regression models with an interaction term between attendance group and treatment allocation group, as well as separately as an additional continuous variable in Cox proportional hazards regression modelling.
iii) Baseline ferritin threshold (≤1200µg/L and > 1200µg/L)
iv) According to presence/absence of comorbidities (diabetes, cardiovascular and cerebrovascular disease, lung disease).
Methods for any additional analysis {20b}: Factors associated with high ferritin: associations at baseline will be explored using both ferritin as a continuous outcome and as a categorical dichotomous outcome (high and not high) using multivariable linear and log binomial regression models respectively. Factors considered for associations with the outcome are serum hepcidin, TSAT, soluble transferrin receptor, percentage hypochromic red cells, reticulocyte haemoglobin content, liver iron, CRP, tumour necrosis factor alpha, IL1, IL6, infection rates, CV events, hepatitis, medications, comorbidities, dialysis vintage, dialysis access, hospitalisations, time variation of ferritin. Only variables significant at a nominal level α= 0.1 in univariable models will be included in a multivariable model. Variables will then be excluded from the multivariable models according to a stepwise backward selection method and a final multivariable model identified that include only variables with a p<0.1. Variables in the multivariable models will be considered to have an independent significant statistical association with the outcome if their p-value is <0.01, variables with a p value between 0.1 and 0.01 will be considered border-line significant/show a weak signal of association with the outcome.
Assessment of the comparability of the current ferritin assay platforms: ferritin values assayed using the Abbott ARCHITECT and the Vitros® ortho XT7600 platforms will be compared using a one sample paired t-test. Bland-Altman plots and Passing and Bablok regression for comparing two methods will be used to assess agreement, constant disagreement or proportional disagreement of results obtained using the two platforms.
Health economic analysis will also be carried out, using the primary outcome measures for the trial to inform a modelling study. We will borrow cost and quality of life estimates from other studies/data sources.
Definition of analysis population relating to protocol non-adherence {20c}: Both primary and secondary endpoints will be analysed according to the intention to treat principles (All participants data will be analysed according to the treatment allocation, regardless of what treatment they received and either they completed the follow up or not). A secondary per-protocol analysis of all endpoints will be conducted. The per-protocol population will be all those who completed the treatment as allocated in the 2 groups.
Methods: Monitoring
Data safety and monitoring board (DSMB) {21a}: An independent DSMB (none of whom will be chief investigators or associate investigators) has been established to review the progress of the study and monitor adherence to the protocol, participant recruitment, outcomes, complications, and other issues related to participant safety. They will also monitor the assumptions underlying sample size calculations for the study and alert the investigators if they see substantial departures from the assumptions as the data accumulate.
The DSMB includes two clinician-researchers familiar with clinical trials, a statistician, and an Indigenous clinical researcher. The DSMB met before recruitment commenced and 6 month following commencement of recruitment and will continue to meet at annual intervals and at its conclusion. Progress reports documenting serious adverse events will be provided to the DSMB 6-monthly. We have planned a formal interim analysis.
The DSMB will make recommendations as to whether the study should continue or be terminated, consider participant safety or other circumstances as grounds for early termination, including either compelling internal or external evidence of treatment differences or feasibility of addressing the study hypotheses (e.g., poor participant enrolment, poor adherence).
First Nation (Aboriginal and Torres Strait Islander) Reference Groups (FNRG): Two First Nation Reference Groups (FNRG) have been established to provide oversight regarding culturally appropriate conduct of research in their respective HREC approved jurisdictions. Each FNRG consists of 7-10 members, of whom all are First Nations. One member each is an independent First Nation academic, with two (2) members from each of the dialysis units: FNRG 1) Top End – Royal Darwin Hospital, Nightcliff, Palmerston, Katherine and Tiwi Islands. FRRG 2) Central Australia - Alice Springs Hospital, Flynn Drive, Gap Road and Tenant Creek.
Of the two members from each site, ideally one will be male, and one will be female. The members can self-nominate or be nominated by or from First Nation communities and community-controlled services which are partners in this study. The FNRG will meet regularly throughout the period of the research conduct, and a representative will join the Trial Management Committee. If requested by the FNRG, the INFERR Clinical Trial Manager or the CI will attend the meetings to provide study progress reports.
Trial co-ordination teams: This trial is co-ordinated from the Menzies School of Health Research in Darwin, but a coordination team is set up in Alice Springs. The teams coordinate the day to day running of the trial and meets weekly and work in collaboration with all the other investigators and research staff.
Trial Management Committee (TMC): The trial management committee includes a chair, co-chair, all chief investigators and associate investigators, the clinical trial manager, research staff and chairs, or proxy of each FNRG. This committee is the steering committee of the trial responsible for all aspects of the trial and making sure the trial is on course.
Interim analyses and stopping guidelines {21b}: The DSMB will conduct an interim analysis after 288 patients have been randomised (i.e., at 50% recruitment) and followed for 24 Months (2 years) following the date of the first patient randomised.
The interim analysis will review outcome data and answer the following questions:
- Are there any significant safety issues that may present an ethical issue in continuing the study? This may include adverse events, but also study conduct and protocol violations.
- Is there overwhelming data suggesting the superiority of one arm that may present an ethical issue in continuing the study?
- Using the Haybittle-Peto rule, and the differences according to treatment allocation in risk of hospitalisation with all-cause infection or death as the outcome of interest, the study will be stopped early if there are differences according to treatment allocation in risk of hospitalisation with all-cause infection or death rate with a p-value of less than or equal to 0.001.
- Are there any other factors that may impact on the feasibility / usefulness of the study? E.g., rate of enrolment, unexpected low rate of outcomes, unable to fund, protocol violations etc.
Harms
Adverse events (AE) and reporting {22}
Serious adverse events (SAEs): A SAE is defined as any experience that:
- Results in death
- Is life-threatening.
- The term “life threatening” refers to an event in which the participant was at risk of death at the time of the event. It does not refer to an event, which hypothetically may have caused death, if it were more serious.
- Results in unexpected prolongation of existing hospitalisation
- Results in persistent or significant disability/incapacity
- Is a medically important event or reaction.
All SAEs that are considered related to the study drugs require reporting on the SAE form. If the SAE is attributable to disease progression, then this does not require expedited reporting in this trial, even when death is the outcome. The site PI is required to report any SAEs that occur at their site to the approving ethics committee in accordance with local guidelines, in addition the site PI must adhere to any local institutional reporting requirements and study sponsor. Safety reporting will be in line with the National Health and Medical Research Council’s (NHMRC) Guidance: Safety monitoring and reporting in clinical trials involving therapeutic goods (2016) (53).
Suspected Unexpected Serious Adverse Reactions (SUSAR): An adverse event that is both serious and unexpected (not listed in the product information and not attributable to disease progression) and related to the study drug meets the definition of a SUSAR. All SUSARs must be entered on the SAE CRF and reported to the sponsor or delegate within 24hours from the time of the site study team becoming aware of it. If the SUSAR is fatal or life-threatening the Sponsor will report to the TGA within 7 calendar days of becoming aware and any follow-up reports required within a further 8 calendar days, for all others 15 calendar days of becoming aware.
The site PI is required to report any SUSARs that occurs at their site to the approving ethics committee in accordance with local guidelines, in addition the site PI must adhere to any local institutional reporting requirements. Safety reporting will be in line with the NHMRC’s Guidance: Safety monitoring and reporting in clinical trials involving therapeutic goods (2016)(53).
Adverse drug reactions (ADRs): Investigators will be asked to report all suspected adverse drug reactions (regardless of severity or seriousness) which are thought to be related to study drugs in both intervention and control arms within the first 24 hours post study drug infusion. These data will be collected routinely on CRFs.
Causality
Assessment of Causality for Study Drugs: The Principal investigator or their delegate is responsible for assessing the relationship to study drugs using clinical judgment and the following considerations:
Unrelated: Evidence exists that the adverse event has an aetiology other than the study drugs. For SAEs, an alternative causality must be provided (e.g., pre-existing condition, underlying disease, intercurrent illness, or concomitant medication).
Related: There is reasonable possibility that the event may have been caused by the
Study drugs. It should be emphasized that ineffective treatment should not be considered as causally related in the context of adverse event reporting.
The degree of certainty with which an adverse event is attributable to treatment, or an alternative cause will be determined by how well the event can be understood in terms of:
- Temporal relationship with the administration of the treatment or cessation of treatment
- Reactions of a similar nature previously observed in the individual or others following treatment.
The relationship of the adverse event to treatment will be specified as follows:
Not related In the PI’s opinion, there is not a causal relationship.
Unlikely The temporal association between treatment and the adverse event is such that treatment is not likely to have any reasonable association.
Possibly The adverse event could have been caused by treatment.
Probably The adverse event follows a temporal sequence from the time of treatment and cannot be reasonably explained by the known characteristics of the participant’s clinical presentation/history.
Definitely The adverse event follows a reasonable temporal sequence from the time of treatment or reappears when the treatment is repeated.
Non-expedited reporting of adverse events and adverse drug reactions: In addition to the expedited reporting described above, a summary of all adverse drug reactions (to any of the study drugs including SAEs, will be reported to the DSMB and HREC as per local guidelines.
Summary of expedited reporting of adverse events and adverse drug reactions: In summary: SAEs not thought to be related to the study drug do not need to be reported in this trial, however this data will be collected for outcome measures. SAEs thought to be possibly, probably or definitely related to study drug (e.g., an anaphylactic reaction to IV iron) must be reported by the PI or CI, research nurse or their delegate to the sponsor (in this case the Clinical Trial Manager and CIs at Menzies) within 24 hours of becoming aware of the event. Reporting to the HREC will be in accordance with local guidelines. If it is an expected side effect (i.e., one listed in the product information, such as allergic reaction or diarrhoea), it does not need to be reported to the TGA. If it is both unexpected and serious, it needs to be reported to the TGA within 7 days (fatal or life threatening) or 15 days (other).
Auditing {23}
Study monitoring will be provided by the responsible monitor(s) at the Menzies School of Health Research (or designee) in accordance with the Monitoring Plan and "International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use" Good Clinical Practice.
The responsible monitor will visit each study site at least once per year and will be allowed, on request, to inspect the various records (source documents, paper CRFs, eCRFs and other pertinent data) provided that participant confidentiality is maintained in accord with local requirements.
It will be the monitor's responsibility to inspect the eCRFs throughout the study, to verify the adherence to the protocol and the completeness, consistency and accuracy of the data being entered on them. The monitor must verify that the participant received the study drug as randomised. The monitor should have access to laboratory test reports and other participant records needed to verify the entries on the eCRF. The investigators and site staff and teams agree to cooperate with the monitor to ensure that any problems detected in the course of these monitoring visits are resolved in a timely manner.
Safety: All the trial medications are licensed for use in Australia with established safety profiles and routinely used in clinical practice. A safety measure was written into the protocol to avoid putting participants at risk of either excessive or insufficient IV iron administration according to the current standards for haemodialysis patients. Prior to receiving any study treatment participants blood results are reviewed for ferritin and TSAT levels, participants treatment varies based on the results of these blood levels:
- Assigned study drug will be administered for ferritin levels between ≥ 700µg/L & ≤ 2000µg/L and TSAT <40% and ferritin <700µg/L and TSAT <40% (Arm A) and no treatment for ferritin levels between ≥700µg/L & ≤2000µg/L and TSAT <40% (Arm B) and rescue therapy for ferritin levels <700µg/L and TSAT<40% (both arms)
- For both arm A and B, if ferritin levels are <700µg/L and TSAT <40% - ‘rescue treatment’ will be administered for that month.
- Ferritin > 2000μg/L and/or TSAT ≥40% for both arm A and B- study treatment will be withheld and receive no treatment for that month.
Ethical and dissemination
General ethical considerations
The study is conducted according to the declaration of Helsinki, the Australian National Health and Medical Research Council (NHMRC) criteria for the ethical conduct of research in humans and the principles of Good Clinical Practice (54).
All drugs in this study are registered for use in Australia. Both treatments are unlikely to cause harm and have been proven safe as they are used daily in routine clinical practice. Written informed consent will be sought from all participants.
Research ethics approval {24}
Approval has been awarded the relevant human research ethics committees (HRECs) for all sites (The Human Research Ethics Committee of the Northern Territory Department of Health and Menzies School of Health Research (HREC) Reference Number: 2019-3536 and Central Australia Human Research Committee (CAHREC); Reference Number: CA-19-3567).
The study protocol, patient information sheet, participant consent forms, and any other documents or media required for ethics approval were submitted to the relevant Human Research Ethics Committees for approval before the study commenced. Each HREC reviewing the protocol must be properly constituted as per regulatory requirements (according to NHMRC requirements) and have the capacity to review the study. Approvals must specify the study title, version numbers, and identify all documents reviewed and state the date of review. No amendments to, or deviations from, the protocol must be initiated without prior written approval from the relevant HREC. The exceptions to this are:
- administrative aspects that have no bearing on participants.
- the need to address regulatory requirements; and/or,
- the need to eliminate immediate hazards to the participants.
Protocol amendments {25}
The investigator will inform the HRECs of the following:
- all protocol amendments, informed consent changes or revisions of other documents originally submitted for review.
- serious and/or unexpected adverse events attributable to study drugs
- new information that may affect the safety of the participants or the proper conduct of the trial.
- annual updates of study progress
- termination of the study including provision of a final study report.
Consent or assent {26}
Informed consent {26a}: An informed consent discussion will be held with each participant. The consent process will be carried out by a study team member delegated informed consent responsibility by the PI with the assistance of an Aboriginal research assistant. The information for the discussion will be provided in written and oral formats that have been approved by the HREC and in a language comprehensible to the potential participant, using interpreters if the teach back method is unsuccessful.
The information presented will detail the exact nature of the trial and what is expected of the participant including any risks or benefits in taking part. It will be clearly stated that the participant is free to withdraw from the trial at any time for any reason without prejudice to future care, and with no obligation to give the reason for withdrawal. The participant will be allowed ample time to read or have read to the Patient Information Sheet and ask questions.
The participant will personally sign and date the latest approved version of the consent form, as will the investigator or the delegated study team member (who conducted the consent discussion). If one was used, an interpreter will also sign and date the consent form. Where the participant is illiterate, they can sign the consent form with their mark rather than their signature, a witness must be present during the entire consenting process and must be able and willing to sign the consent form also. A copy of the participant information sheet and consent form will be provided to the participant. No trial related procedure will be undertaken before documented informed consent is obtained. The original copy of the consent form will be retained in the Investigator Site File (ISF) at the recruitment site and a copy placed in the patient’s medical records. The person conducting the consent discussion will document this in the participant’s medical record and enter data into the Day 1 CRF
Confidentiality {27}
The trial management committee will be the custodians of the final trial dataset. No-one outside the trial management committee will be given access to the data without the permission of the trial management committee. No identifying data will be given to any third parties at any stage. Following study close out and locking of the database, it will be stored on the servers of the Menzies School of Health Research.
Dissemination policy {31}
Communicating trial results {31a}: The trial results will be communicated to all investigators and to IRG members prior to publication or presentation. The trial results will be presented at national and international scientific conferences. The trial results will also be submitted for publication to a peer reviewed scientific journal, irrespective of the results. The FNRG will advise on participant and community feedback content and method of deliver. A plain-language summary of the trial results will be made available to individual participants.
Authorship eligibility {31b}: The first draft of the manuscript will be written by CIs Majoni, Cass and Lawton, and subsequent drafts will have input from the rest of the Trial Management Committee (meaning all named Chief Investigators and Associate Investigators.) The decision where to publish will be made by the Trial Management Committee. The authorship of the paper will include all of the Trial Management Committee who meet ICJME criteria for authorship. Contributions of other study participants will be recognized by the following language at the end of the named authors’ list: “. . .and INFERR study group”. The INFERR study group will consist of all named investigators and will be listed in the collaborators section of the paper.
Regulatory approvals
Even though all products are licensed for use in Australia, IV iron will be used outside the current guideline recommendations. Hence a Clinical Trials Notification (CTN) has been lodged with the Therapeutic Goods Administration (TGA).
{kind=link}