Mice and genotyping
Genotypic identification of transgenic mice was carried out according to the method described in published literature[26]. All animal experiments were performed in accordance with the protocols approved by the Animal Care and Use Committee of Southeast University and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Auditory brainstem response (ABR)
A TDT System III workstation running SigGen32 software (Tucker-Davis Technologies, USA) was used to record ABRs as previously described[27, 28]. The mice were anesthetized by intraperitoneal injection of 0.01 g/ml pentobarbital sodium (100 mg/kg body weight). After deep anesthesia, three fine-needle electrodes were inserted under the skin of the mouse at the vertex of the skull, behind the tested ear, and on the back near the tail. The mice were then put into a soundproof room for the ABR test. The TDT hardware and software (BioSig and SigGen) were used to generate the acoustic signals and to process the responses. The ABRs were elicited with tone bursts at 4, 8, 12, 16, 24, and 32 kHz. The tests were performed at a 5 dB interval from 90 dB to 10 dB at each frequency with a gradually decreasing intensity, and ABR thresholds were recorded as the lowest sound intensity at which a stable wave III could be seen and repeated.
Immunohistochemistry
The basilar membranes of the newborn mouse cochleae were dissected with microsurgery forceps and incubated with 4% paraformaldehyde for 1 h at room temperature (RT), while the basilar membranes from ossified cochleae were carefully dissected with a microsurgery scalpel after incubating in 4% paraformaldehyde and 0.5 M EDTA overnight. Whole mounts of the basilar membrane were then blocked with 10% heat-inactivated donkey serum, 1% bovine serum albumin (BSA), and 1% Triton X-100 in PBS (0.1 M phosphate buffer, pH 7.2) for 1 h at RT. The samples were incubated with primary antibodies diluted in 5% heat-inactivated donkey serum, 1% BSA, and 10% Triton X-100 overnight at 4°C. The tissues were washed three times with PBST (PBS and 1% Triton X-100) and further incubated at RT for 1 h with secondary antibodies (Alexa Fluor 647 or 555 or 488, Invitrogen) diluted in 0.1% BSA and 0.1% Triton X-100. Finally, the tissues were again washed with PBST three times and mounted on a slide. A Zeiss LSM700 confocal microscope was used to take images.
The primary antibodies used in the experiment were FAM73a (Biobyt, orb187931), FAM73b (Novusbio, NBP1-86701), Myosin7a (Proteus Bioscience, 25-6790; DSHB, 138-1); 3-NT (Sigma, N5538), 4-HNE (Abcam, ab46545), F4/80 (Abcam, ab6640); Iba1 (Wako Pure Chemicals, 019-19741), IL-12 (Abcam, ab210255), MHCII (Abcam, ab23990), CD4 (Santa Cruz Biotechnology, sc-20079), IRF1 (Santa Cruz Biotechnology, sc-514544), and Parkin (Santa Cruz Biotechnology, sc-32282). A TUNEL kit (Vazyme, A112-03) was used to detect apoptotic cells according to the manufacturer’s instructions.
qRT-PCR
Total RNA from the brain, from different parts of the cochlea, and from whole cochleae was extracted with ExTrizol Reagent (Protein Biotechnology, PR910), and the reverse transcription from mRNA to cDNA was carried out using cDNA Synthesis kits (Thermo Fisher Scientific, K1622) according to the manufacturer’s instructions. The qPCR was performed using an Applied Biosystems CFX96 qPCR system (Bio-Rad, Hercules, CA, USA) and the SYBR Green (Rox) qPCR Master Mix (Roche Life Science, 04913850001). Validated primers were designed for targeted DNA or mRNA sequences (Table1). The qPCR protocol was an initial denaturing step of 15 s at 95°C followed by 40 cycles of 15 s denaturation at 95°C, 60 s annealing at 60°C, and 20 s extension at 72°C. The expression of mRNA was normalized using the values of Gapdh, and the results were analyzed using the comparative cycle threshold (ΔΔCt) method.
Western blot
Cochleae from two mice were dissected in cold PBS and lysed with 150 ml RIPA Lysis Buffer (Medium, Hangzhou Fu De Biological Technology) and 3 µl 50× protease inhibitor cocktail (Hangzhou Fu De Biological Technology) at 4°C. The primary antibodies were detected by HRP-conjugated secondary antibodies using the ECL detection system. The western blot bands were semiquantified using ImageJ software, and the band densities were normalized to background and the relative optical density ratio was calculated by comparison to the reference protein GAPDH or β-actin.
Each experiment was repeated at least three times. The following primary antibodies were used: FAM73a (Abcam, ab121532), FAM73b (Santa Cruz Biotechnology, sc-20437), 3-NT (Sigma, N5538), Arg1 (Abcam, ab239731), IRF1 (Santa Cruz Biotechnology, sc-514544), Parkin (Santa Cruz Biotechnology, sc-32282), CHIP (Santa Cruz Biotechnology, sc-133066), and ubiquitin (Santa Cruz Biotechnology, sc-8017).
Immunoprecipitation (IP)
Cochleae from two mice were lysed with 150 ml RIPA lysis buffer (Medium, Hangzhou Fu De Biological Technology). CHIP was isolated with antibodies targeting CHIP (Santa Cruz Biotechnology, sc-133066), and Protein A+G was used to capture the antibodies. The ubiquitinated proteins were detected by western blot using anti-ubiquitin antibodies (Santa Cruz Biotechnology, sc-8017).
Scanning electron microscopy
The cochlear specimens from P30 mice were collected and immediately fixed in 2.5% glutaraldehyde (Sigma-Aldrich, G5882) diluted in 0.1 M phosphate buffer (pH 7.2) for 24 h at 4°C. The tissues were then decalcified for 3 h in 0.5 M EDTA, post-fixed for 2 h at 4°C in 1% osmium tetroxide, dehydrated in ethanol, and embedded in araldite CY 212 (TAAB, E009). The ultrathin sections were stained with alcoholic uranyl acetate (Polysciences, 6159–44–0) and alkaline lead citrate (SigmaAldrich, 15326), washed gently with distilled water, and imaged with a JEM 1230 transmission electron microscope (JEOL Ltd, Tokyo, Japan).
Fluorescence intensity measurement
Different groups of cochleae were fixed, labeled with the same solution, and processed in parallel. The tissue was photographed with confocal microscope using the same parameters. The immunolabeling intensity of antibodies was measured using ImageJ software in which a region of interest was drawn and the mean gray value intensities were measured from 4 or 5 sections per cochlea.
The number and morphology of basilar membrane macrophages
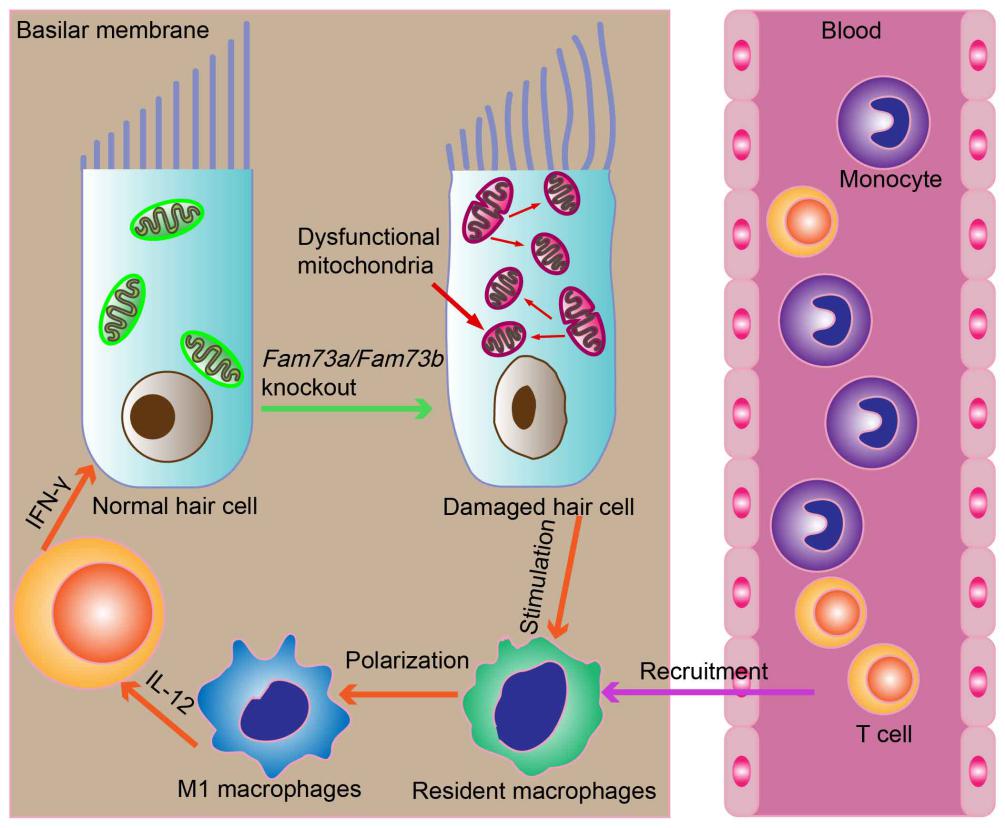
Macrophages were distributed throughout the basilar membrane, and exhibited dendritic, irregular, amoeboid, and spherical morphology. They were identified with surface markers F4/80 and Iba1 as have been used in previous studies[29]. To assess the number of macrophages in the apical, middle, and basal turns of the basilar membrane, F4/80-labeled macrophages were counted under the confocal microscope. Images at 20× magnification taken from each turn of the cochlear whole mounts were used as representative figures. To measure the size of macrophages, ImageJ software was used to outline the membrane boundaries of each cell and calculated the area contained in the drawn region. Five typical cells were selected from each turn of tissue specimen, and their average area represented the size of apical, middle, and basal turns macrophages in the basilar membrane of each individual cochlea.
Macrophage phagocytosis
PHrodo® zymosan bioparticles conjugate (Invitrogen, P35365) was used to evaluate the phagocytic activity of macrophages. The fluorescence of the pHrodo® dye was activated when the zymosan bioparticles were ingested and exposed to a more acidic PH within the acidic phagocytic vacuoles. Because the extracellular pH is more alkaline, bioparticle fluorescence was absent outside the cell. The cochleae were dissected from the skull and placed in live cell imaging solution (A14291DJ, Invitrogen). The membrane labyrinth was opened from the top of the cochlea to remove the basilar membrane, modiolus, and the lateral wall tissue, thereby exposing the inner surface of lateral wall of scala tympani at the basal turn of the cochlea. Then the basal turn was divided into several pieces so that the cochlear bone wall could be flat on the slide. PHrodo® zymosan bioparticles® conjugate incubated the collected tissues for 90 minutes at 37°C, and then they were rinsed three times for 5 min each using live cell imaging solution. 4% buffered formalin fixed the collected tissues for 4 hours and then EDTA decalcified at 4°C for 1 day. Subsequently, the primary antibody against F4/80 and the appropriate secondary antibody incubated the collected tissues to visualize macrophages.
Drug administration
Clophosome®-A - Clodronate Liposomes (LCCA) (FormuMax, F70101C-A) provide superior efficiency of macrophage depletion. We intraperitoneally injected mice with LCCA every other day from P30 to P60 at 70 mg/kg. We collected and dissected the cochleae and measured HC loss and the number of macrophages when the drug administration was completed.
Statistical analysis
Microsoft Excel and GraphPad Prism software were used for statistical analyses. All of the data are presented as mean ± SD, and all experiments were repeated at least three times. Two-tailed, unpaired Student’s t-tests were performed. P-values <0.05 were considered significant, and the level of significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001. All statistical tests were justified as appropriate, and the data met the assumptions of the tests. The variance was similar between the statistically compared groups.
{kind=link}