Macrophage Membrane Derivation
Murine macrophage RAW264.7 cells, kindly provided by Stem Cell Bank (Chinese Academy of Sciences, China), were cultured in Dulbecco’s modified Eagle’s medium (Hyclone, USA) containing 10% fetal bovine serum (Gibco, Australia) and 1% penicillin/streptomycin (Gibco, USA) at 37 °C in 5% CO2. The membrane was extracted from RAW264.7 cells using a membrane protein extraction kit (Beyotime, China). First, the cells were immersed for 15 min in ice-cold membrane protein extraction reagent; thereafter, cells were moderately disrupted using a Dounce homogenizer. Nuclei and a small number of unbroken cells were removed by low-speed centrifugation (700 × g, 10 min), and the supernatant was subjected to high-speed centrifugation (14000 × g, 30 min) to obtain the cell membrane precipitate. The membrane protein content was measured using a BCA kit (Beyotime). To obtain macrophage membrane vesicles, an ultrasound bath (42 kHz, 100 W) was used. Ultrasound was applied to cell membranes for 15 min, followed by 11 extrusions using an Avanti mini extruder with 400 nm polycarbonate porous membranes (Avanti, Canada).[34]
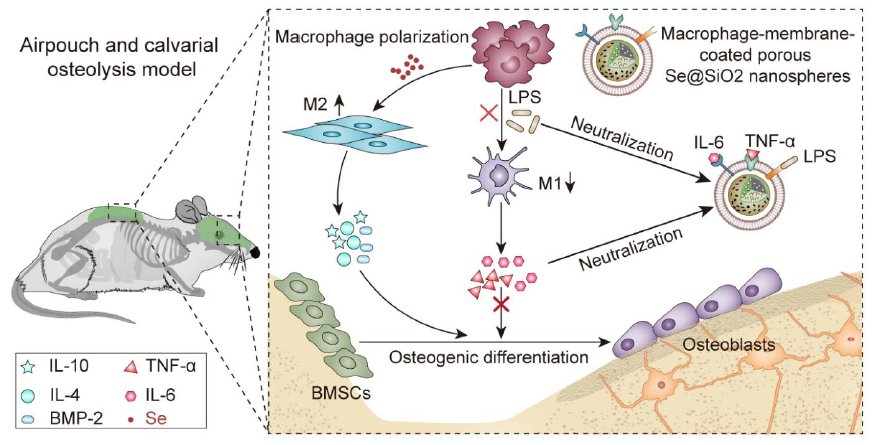
M-Se@SiO2 Preparation and Characterization:
Porous Se@SiO2 nanospheres were synthesized according to our previous study [36]. First, Cu2−xSe nanocrystals were oxidized to form Se quantum dots. Solid Se@SiO2 nanospheres were formed by coating silica onto Se quantum dots by orthosilicate hydrolysis in an alkaline environment. Next, the solid Se@SiO2 nanospheres were coated with polyvinylpyrrolidone and treated with hot water to form porous structures. TEM (TF20; FEI, USA) and X-ray diffractometry (Rigaku, Japan) were used for nanoparticle detection and characterization. The collected macrophage membranes and porous Se@SiO2 nanospheres were mixed at a 1:1 mass ratio of membrane proteins to nanoparticles using the ultrasound bath for 3 min. The membranes were then passed 11 times through 100 nm polycarbonate porous membranes using the Avestin mini extruder to obtain macrophage-membrane-coated porous Se@SiO2 nanospheres (M-Se@SiO2). M-Se@SiO2 were detected by negative staining in TEM. Briefly, 3 μL of nanoparticle suspension (1 mg/mL) was deposited on a copper grid and subsequently stained with 1 wt% phosphotungstic acid. The samples were then observed using a Talos 120 kV Sphera microscope. Dynamic light scattering was used to measure the size and zeta potential of the nanoparticles. M-Se@SiO2 at 1 mg/mL was mixed with 2× phosphate-buffered saline (PBS) at a 1:1 volume ratio, and its stability in PBS was assessed. The Se release from porous Se@SiO2 nanospheres and M-Se@SiO2 was studied separately using a Leeman ICP-AES Prodigy instrument as previously described.[49]
LPS and Cytokine Neutralization
To evaluate the LPS- and cytokine binding ability of M-Se@SiO2, M-Se@SiO2 nanospheres (1 mg/mL) were mixed with PBS containing 10% fetal bovine serum (FBS) and fluorescein isothiocyanate (FITC)-LPS (Sigma, 100 ng/mL), TNF-α (85 pg/mL), or IL-6 (360 pg/mL) at 37 °C for 30 min. The samples were centrifuged at 16,000 × g for 15 min to remove the nanospheres. LPS remaining in the supernatant was measured as the fluorescence intensity, and cytokines in the supernatant were quantified by ELISA (Anogen, Canada).
Characterization of Membrane Proteins
Membrane proteins from macrophages were extracted using a membrane protein extraction kit (Beyotime) and the abundance of cell membrane surface proteins, including TLR4, TNFR1, and IL6-R, was detected by western blotting.
BMDM Cultures
The femurs of C57/BL6 mice were washed with α-modified Eagle’s medium (α-MEM; Gibco, USA) to obtain bone marrow cells. The extracted cells were maintained in complete α-MEM containing 10% FBS (Gibco, Australia), 1% penicillin/streptomycin (Gibco, USA), and 40 ng/mL macrophage colony-stimulating factor (Peprotech, USA) at 37 °C in 5% CO2 for 7 d to obtain mouse BMDMs. The BMDMs were than seeded into cell culture plates and treated with LPS (Sigma, 100 ng/mL), with or without porous Se@SiO2 nanospheres (Se@SiO2 group; 10 µg/mL) or M-Se@SiO2 (M-Se@SiO2 group; 10 µg/mL).
Cell Biocompatibility Assay
Cell viability was analyzed using a Cell Counting Kit 8 (CCK-8; Dojindo, Japan). BMDMs were seeded in 24-well plates at a density of 5 × 105 per well, whereas BMSCs were plated at a density of 1 × 105 per well. After 24 h of incubation, the cells were cultured in medium containing 10% CCK-8 for 2 h at 37 °C. The absorbance was then read at 450 nm using a microplate reader (BioTek, USA)
Immunofluorescence Staining
Immunofluorescence staining of the M1-like macrophage marker CCR7 and M2-like macrophage marker ARG1 was performed to assess macrophage polarization. After 24 h of culture, cells were fixed in paraformaldehyde (4%), blocked with Blocking Buffer for Immunol Staining (Beyotime, China) for 15 min, and incubated with mouse anti-CCR7 (1:100, Abcam, USA) or rabbit anti-ARG1 (1:100, Abcam, USA) antibodies overnight at 4 °C. The next day, the cells were washed and incubated with donkey anti-mouse Alexa Fluor 594 (1:200, Abcam, USA) or donkey anti-rabbit Alexa Fluor 488 (1:200, Abcam, USA) for 1 h at room temperature protected from light. The cells were then washed with PBS and the nuclei were stained with 4ʹ,6-dimidazole-2-phenylindole (DAPI) for 5 min. Images were taken using a DM8 microscope (Leica, USA).
Flow Cytometry
Flow cytometry was carried out to analyze the abundance of the M1 marker CCR7, M2 marker CD206, and general macrophage marker F4/80. After 24 h of culture, the cells were scraped, washed, blocked for 15 min with Blocking Buffer (Beyotime), and finally stained for 1 h with allophycocyanin (APC)-conjugated anti-CCR7 antibody (1:100, BioLegend, USA) or FITC-conjugated anti-CD206 antibody (1:100, BioLegend, USA). PE-conjugated anti-F4/80 (1:100, BioLegend, USA) was used to label all macrophages. The cells were analyzed using BD flow cytometry with the FlowJo software
Enzyme-Linked Immunosorbent Assays
After 24 h of incubation, the cell culture medium was collected and centrifuged. Cytokine levels (TNF-α, IL-4, IL-6, and IL-10) in the supernatant were determined using ELISA kits (Anogen, Canada) according to the manufacturer’s instructions
Real-Time Polymerase Chain Reaction
After 24 h of incubation, the total RNA was extracted using an RNA extraction column kit (EZBioscience, USA) according to the manufacturer’s instructions. Subsequently, complementary DNA was synthesized from 1 μg of total RNA using a cDNA synthesis kit (EZBioscience, USA). Quantitative RT-PCR was performed using a SYBR Green Master mix (EZBioscience, USA) on LightCycler 480 (Roche, USA). The primers used are shown in Table S1.
Western Blotting
The cells were pretreated with Se@SiO2 or M-Se@SiO2 for 1 h, and then LPS was added (100 ng/mL). After 30 min, the cells were lysed with RIPA lysis buffer containing protease and phosphatase inhibitors (EpiZyme, China). The extracted total proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene fluoride membranes. The membranes were blocked in blocking solution (Beyotime) for 15 min, washed, and incubated with primary antibodies overnight at 4 °C. p38/p-p38, extracellular signal-regulated kinase (ERK)/p-ERK, and p65/p-p65 antibodies (1:1000, Cell Signaling, USA) were used for the experiments. After washing, the membranes were incubated with goat anti-rabbit IgG-horseradish peroxidase (1:1000, Cell Signaling, USA) for 1 h at room temperature. Blots were developed with enhanced chemiluminescent reagent (Millipore, USA) and Tanon Imaging System (Tanon, China) for chemiluminescent detection. Relative band intensities were quantified using ImageJ software, and all results were normalized to β-actin expression.
BMSC Cultures and Conditioned Medium Preparation
Primary mouse BMSCs were isolated as previously described[4] and cultured in α-MEM containing 10% FBS and 1% penicillin/streptomycin. After culturing BMDMs for 24 h, media from the control, LPS, LPS+Se@SiO2 and LPS+M-Se@SiO2 group cultures were collected and centrifuged; the resulting supernatant was then mixed with osteogenic induction medium (Cygen, China) at a ratio of 1:2 to obtain the conditioned medium. Next, BMSCs were seeded into 24-well plates at a density of 2.5 × 104 cells per well. Following cell adherence, the culture medium was replaced with conditioned medium, which was subsequently changed every two days
Alkaline Phosphatase (ALP) and Alizarin Red (ARS) Staining
BMSCs were fixed using 4% paraformaldehyde and stained with ALP (Beyotime, China) or ARS (Cytogen, China) dyes after 14 d of culture. ALP was quantified using an ALP assay kit (Beyotime, China) according to the manufacturer’s instructions. Quantitative analysis of ARS staining was performed by adding 10% acetylchlorinated pyridine to release ARS after ARS staining and subsequently by measuring the optical density at 600 nm.
Analysis of Gene Expression in BMSCs Exposed to BMDM-Conditioned Medium
After 2 weeks of incubation, the expression of the osteogenesis-related genes of bone morphogenetic protein-2 (BMP-2), osteocalcin (OCN), and osteopontin (OPN) was analyzed by RT-PCR. The primers used are shown in Table S1
In Vivo Air Pouch Model in Mice
The animal experiments were approved by the Animal Care Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. C57BL/6 mice were used in the experiments as described previously.[5] After mild anesthesia of the mice using pentobarbital, 5 mL of sterile air was injected subcutaneously. Four days after air pouch formation, 0.5 mL PBS or 0.5 mL PBS with 1 µg/mL LPS was injected with or without porous Se@SiO2 or M-Se@SiO2. Four days after injection, the mice were sacrificed, and exudates obtained by washing the air pouches with 2 mL PBS were centrifuged and stored at −80 °C for ELISA. Finally, the air pouch tissue was collected and fixed with 4% paraformaldehyde. Tissue sections were subjected to hematoxylin and eosin (H&E) or Masson trichrome staining to assess inflammation, immune cell infiltration, and membrane thickness. Immunofluorescence staining was used to detect CCR7- and ARG1-positive cells in the air pouch tissue. Images were captured using a DM6 microscope (Leica, USA) and analyzed using the ImageJ software.
In Vivo Calvarial Osteolysis Model in Mice
Mice were anesthetized with intraperitoneal pentobarbital injections. A 1-cm long incision was made in the middle of the cranium, and the cranial periosteum was separated from the calvarium. Next, 50 µL of LPS (1 mg/mL) was embedded under the periosteum around the sagittal midline suture of the calvaria. In the experimental groups, PBS, porous Se@SiO2, or M-Se@SiO2 were injected intraperitoneally at a dose of 0.5 mg/kg every 3 d. The animals were sacrificed 14 d after surgery. The cranial bones were carefully harvested, fixed in 4% paraformaldehyde, and stored in 70% ethanol until they were imaged with a micro-CT scanner (Bruker micro-CT) at a resolution of 18 mm. Bone volume to tissue volume ratio (BV/TV), total porosity, and the number of pores were measured and analyzed using CT Analyser Software (Bruker) as previously described.[50] Before to paraffin embedding, bones were decalcified in 14% EDTA, pH 7.4 for 2 weeks. Thereafter, H&E Masson trichrome and tartrate-resistant acid phosphatase (TRAP) staining was performed. The stained sections were imaged using a DM6 microscope (Leica, USA). In addition, the heart, kidney, liver, spleen and lungs of mice were collected and fixed to assess biosafety aspects of the administered nanoparticles in major organs. The sections of major organs were paraffin-embedded, cut, and stained with H&E.
Statistical analysis
Data are presented as mean ± standard deviation and were analyzed with SPSS v. 18.0 software (SPSS Inc., Chicago, IL, USA). Statistical significance among groups was evaluated using the one-way analysis of variance and the t-test. Results with p < 0.05 were considered statistically significant.
{kind=link}