Identification of DEGs
According to GSE121248 dataset analysis, 943 DEGs were successfully identified, including 325 upregulated and 618 downregulated genes. For GSE64041 dataset, 289 DEGs were observed, including 87 upregulated and 202 downregulated genes. For GSE62232 dataset, 1,355 DEGs were identified, involving 817 upregulated and 538 downregulated genes. Venn analysis was performed to examine the intersection among the three DEGs profiles. Then, 197 DEGs were identified from the three profile datasets (Table 1). Obviously, 54 DEGs were significantly upregulated (Fig. 1A), while 143 DEGs were markedly downregulated (Fig. 1B) in HCC tissues. These 197 DEGs were plotted in Fig. 1C, where the red and green dots represented the upregulated and downregulated DEGs, respectively. In addition, the mRNA expression level of these 197 DEGs was visualized in the form of a heatmap using data profile GSE20347 (Additional file 1:Figure S1).
Table 1
The common DEGs of three gene expression profiles (adj. Pval. <0.05, |logFC|>1.0)
| DEGs | Gene symbol |
| Upregulated (54) | SPINK1; TPX2; EDIL3; ASPM; FLVCR1; AKR1B10; GINS1; SRXN1; KPNA2; ANLN; NQO1; FOXM1; EZH2; CCNB2; RBM24; PRC1; CDK1; TOP2A; TXNRD1; SPARCL1; CDC6; FAM72A; MAP2; AURKA; BUB1B; DLGAP5; NMRAL1P1; LEF1; MKI67; CAP2; DTL; GPC3; CCL20; ROBO1; SPP1; SQLE; KIF20A; UBD; RRAGD; CD200; ITGA6; LCN2; MELK; SLC7A11; ITGA2; CCNA2; CDKN3; BUB1; NUF2; NCAPG; UBE2T; CENPF; NUSAP1; ECT2 |
| Downregulated (143) | TUBE1; BBOX1; XDH; SDS; CXCL14; IGF1; DPT; CYP39A1; SLC25A47; PROZ; C8A; ZG16; MBL2; SLC10A1; SLCO1B3; PRG4; CYP1A2; UROC1; FCGR2B; F9; BCO2; ACSM3; CYP2C19; C3P1; LPA; CD5L; GHR; CLEC1B; TAT; LIFR; BHMT; COLEC10; VNN1; LYVE1; STEAP3; SHBG; DNASE1L3; ALDH8A1; NAT2; C7; BCHE; SAA2-SAA4; AKR1D1; CXCL12; GNMT; C1orf168; GPD1; CRHBP; EHD3; WDR72; IDO2; BDH2; CYP3A43; SLC38A4; DBH; FBP1; ADH4; OIT3; MT1M; SLC39A5; CETP; SRD5A2; ADRA1A; PBLD; SRPX; CYP4A22; KLKB1; GNAO1; ENO3; MT1G; SLC19A3; PGLYRP2; TENM1; INS-IGF2; CYP2C8; STEAP4; IL13RA2; SPP2; IGHM; MT1F; FETUB; MFSD2A; HHIP; APOA5; CYP2B7P; KCND3; PPP1R3B; LY6E; ITGA9; OLFML3; CNDP1; FCN3; GBA3; PDGFRA; CLEC4G; PHGDH; CYP2B6; CCBE1; FXYD1; PCK1; KMO; ANK3; CLRN3; MT1H; CLEC4M; NPY1R; ESR1; TDO2; VIPR1; IGFBP3; PLAC8; HAMP; DCN; IL1RAP; RDH16; CYP8B1; TMEM27; AFM; HPGD; LPAL2; THRSP; CYP4A11; STAB2; HGFAC; ADGRG7; OGDHL; PZP; SLCO4C1; FREM2; BMPER; AADAT; GPM6A; HGF; MOGAT2; CYP3A4; EPHX2; GLS2; HABP2; APOF; ANGPTL1; PTGIS; GRAMD1C; SLC7A2 |
Figure 1 Identification of common DEGs from GSE121248, GSE64041, and GSE62232 datasets. Venn diagram of (A) upregulated and (B) downregulated DEGs based on the three GEO datasets. (C) Volcano plot of the 197 DEGs.
Red, upregulation; green, downregulation. The intersecting areas represent
the commonly altered DEGs. The t-test was used to analyze DEGs, with the
cut‑off criteria of |logFC|>1.0 and adj. P < 0.05. DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; logFC, log‑fold change.
Functional enrichment analysis of DEGs
GO annotation and KEGG pathways enrichment analysis were conducted through Enrichr database(http://amp.pharm.mssm.edu/Enrichr/). Figure 2 showed the top 10 enriched GO terms and KEGG pathways. As shown in Fig. 2A, GO BP analysis revealed that these 197 DEGs were significantly enriched in epoxygenase P450 pathway, steroid metabolic process, kynurenine metabolic process and arachidonic acid metabolic process. The top four significantly enriched CC terms included condensed nuclear chromosome, centromeric region, spindle, integral component of plasma membrane, and condensed nuclear chromosome kinetochore (Fig. 2B). For GO MF analysis, the top four significantly enriched terms were oxidoreductase activity, steroid hydroxylase activity, oxidoreductase activity, heme binding (Fig. 2C). Additionally, the top four markedly enriched pathways for these 197 DEGs were Retinol metabolism, Arachidonic acid metabolism, Tryptophan metabolism and Caffeine metabolism (Fig. 2D).
Figure 2 GO annotation and KEGG pathway enrichment analysis of 197 DEGs. The top 10 enriched GO (A) BP, (B) CC, (C) MF terms and (D) KEGG
pathways. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed genes; BP, biological process; CC, cellular component; MF, molecular function.
PPI network construction and hub genes identification
The STRING database was performed to determine the PPI network among
the 197 DEGs. The PPI network including 197 nodes (genes) and 968 edges (interactions) was constructed through STRING database (Additional file 1:Figure S2). The PPI enrichment p-value < 1.0 × 10− 16. Ten genes with the highest degree scores were reged as the hub genes by applying the Cytoscape (v3.6.1) plugin cytoHubba. The results revealed that forkhead box M1(FOXM1) was the hub gene with the highest connectivity degree, followed by aurora kinase A (AURKA), cyclin A2 (CCNA2), cyclin-dependent kinase inhibitor 3 (CCKN3), marker of proliferation Ki-67 (MKI67),enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), cell division cycle 6 (CDC6),cyclin-dependent kinase 1 (CDK1),cyclin B1 (CCNB1),Topoisomerase (DNA) II alpha (TOP2A) (Table 2).
Using cytoHubba software,the PPI network of the screened 10 hub genes were constructed, which had a strong interaction among each other (Fig. 3A). The interaction network of 10 hub genes and their related genes was also identified by the FunRich software (Fig. 3B) [28]. The hub genes and their related genes could be enriched in many biological pathways through the enrichment functions of FunRich tool. KEGG analysis established that markedly enriched pathways for the hub genes included progesterone mediated oocyte maturation, cell cycle, cellular senescence, oocyte meiosis, p53 signaling pathway, Viral carcinogenesis, Lysine degradation, and gap junction (Fig. 3C).
Table 2
Top ten hub genes with higher degree of connectivity.
| Gene symbol | Gene description | Degree |
| FOXM1 | Forkhead box M1 | 36 |
| AURKA | Aurora kinase A | 34 |
| CCNA2 | Cyclin A2 | 34 |
| CDKN3 | Cyclin-dependent kinase inhibitor 3 | 34 |
| MKI67 | Marker of proliferation Ki-67 | 34 |
| EZH2 | Enhancer of zeste 2 polycomb repressive complex 2 subunit | 33 |
| CDC6 | Cell division cycle 6 | 33 |
| CDK1 | Cyclin-dependent kinase 1 | 33 |
| CCNB1 | Cyclin B1 | 33 |
| TOP2A | Topoisomerase (DNA) II alpha | 33 |
Figure 3 Interaction network and KEGG analysis of the hub genes. (A) The top 10 hub genes in the PPI network were screened by Cytoscape (v3.6.1) plugin cytoHubba. The 10 hub genes are displayed from red (high degree value) to yellow (low degree value). (B)The PPI network of the 10 hub genes and their related genes, created by the FunRich software. (C) KEGG pathway enrichment analysis of the 10 hub genes. PPI, protein-protein interaction ; STRING, search tool for the retrieval of interacting genes; KEGG, Kyoto encyclopedia of genes and genomes.
Validation of mRNA expression levels of these 10 hub genes in HCC
First, a differential analysis on the mRNA expression levels of FOXM1, AURKA, CCNA2, CCKN3, MKI67, EZH2, CDK1, CCNB1, and TOP2A, between HCC and non-tumor liver tissues was conducted through the GEPIA database.
As showed in Fig. 4, the mRNA expression levels of (Fig. 4A) FOXM1, (Fig. 4B) AURKA, (Fig. 4C) CCNA2,(Fig. 4D) CCKN3, (Fig. 4E) MKI67, (Fig. 4F) EZH2, (Fig. 4G) CDC6, (Fig. 4H) CDK1, (Fig. 4I)CCNB1, and (Fig. 4J) TOP2A were significantly upregulated in HCC tissues (P < 0.01) compared to those in normal liver tissues. Hierarchical clustering analysis also performed that the mRNA expression levels of all the hub genes were significantly increased in primary hepatic cancer (TCGA Live Cancer, n = 469) tissues compared to non-tumor tissue samples through UCSC Xena Browser (Additional file 1:Figure S3). These findings were consistent with the obtained GEO microarray data.
Figure 4 Validation of the mRNA expression levels of (A) FOXM1, (B) AURKA, (C) CCNA2, (D) CCKN3, (E) MKI67, (F) EZH2, (G) CDC6, (H) CDK1,
(I) CCNB1, and (J) TOP2A in LIHC tissues and normal liver tissues using GEPIA database. These ten box plots are based on 369 LIHC samples (marked in red) and 160 normal samples (marked in gray). *P < 0.01 was considered statistically significant. LIHC, liver hepatocellular carcinoma.
Validation of protein expression levels of these 10 hub genes in HCC
The protein expression levels of these hub genes in HCC were Validated through the HPA database. Obviously, the protein expression levels of (Fig. 5A) FOXM1, (Fig. 5B) AURKA, (Fig. 5C) CCNA2, (Fig. 5D) MKI67, (Fig. 5E) EZH2, (Fig. 5G)CDK1, (Fig. 5H) CCNB1 and (Fig. 5I) TOP2A were not observed in hepatocytes of normal liver tissues, but medium or high expression levels of these hub genes were detected in HCC tissues (Fig. 5A-E and G-I). The low protein expression levels of CDC6 were detected in normal liver tissues, while medium protein expression levels of CDC6 were detected in HCC tissues (Fig. 5F). Unfortunately,the protein expression levels of CDKN3 were not explored because of pending cancer tissue analysis in HPA database. In brief, these present results showed that the mRNA and protein expression levels of the hub genes were overexpressed in HCC tissues.
Figure 5 Representative immunohistochemistry images of (A) FOXM1, (B) AURKA, (C) CCNA2, (D) MKI67, (E) EZH2, (F) CDC6, (G) CDK1, (H) CCNB1, and (I) TOP2A in HCC and non-cancerous liver tissues derived from the HPA database. HCC, hepatocellular carcinoma; HPA, Human Protein Atlas.
Frequencies of genetic alterations and prognostic values of these hub genes.
The frequencies of genetic alterations of these hub genes in LIHC were identified through the cBioPortal database. 32% of LIHC cases showed significant alterations in these 10 hub genes (Fig. 6A). The mRNA upregulation was the most important single alteration for these 10 hub genes in 78 cases (22%) of LIHC. The mRNA expression levels of these 10 hub genes in LIHC were further analyzed. The percentage change in the mRNA expression levels of FOXM1, AURKA, CCNA2, CDKN3, MKI67, EZH2, CDC6, CDK1, CCNB1, and TOP2A in LIHC were 7, 7, 2, 4, 3, 8, 8, 5, 7 and 5%, respectively (Fig. 6B). Furthermore,the relationship between the changes of these hub genes and LIHC prognosis was established through the cBioportal database. Kaplan-Meier plots were performed to compare OS, and FDS/PFS in LIHC patients with or without alterations in the mRNA expression levels of these hub genes. As showed in Fig. 8C, LIHC cases with altered hub gene showed remarkably worse OS relative to those with unaltered hub gene (P = 5.600e-4). LIHC cases with altered hub gene also showed significantly worse DFS (P = 1.940e-3) compared with those with unaltered hub gene (Fig. 6D).
Figure 6 Alteration frequency and prognosis of the 10 hub genes. (A) The summary of the cancer types was used to calculate alteration frequency of the 10 hub genes in LIHC cases. (B) mRNA expression alterations (RNA Seq V2 RSEM) of the 10 hub genes in LIHC patients. (C) OS and (D) DFS/PFS of LIHC patients with altered (red) and unaltered (blue) mRNA expression of the 10 hub genes. LIHC, liver hepatocellular carcinoma; OS, overall survival; DFS/PFS, disease-free survival/progression-free survival.
Survival analysis of the hub genes in HCC
To further explore the relationships between the 10 hub genes and HCC, OS and DFS analysis of the 10 hub genes were performed by Kaplan-Meier plotter, and the GEPIA database. As showed in Fig. 7, the high expression levels of FOXM1, AURKA, CCNA2, CDKN3, MKI67, EZH2, CDC6, CDK1, CCNB1, and TOP2A in LIHC patients were related to poor OS. The unfavorable DFS was also significantly showed in LIHC patients with high expression levels of these 10 hub genes (Fig. 8).
Figure 7 OS of the 10 hub genes overexpressed in patients with liver cancer was analyzed by Kaplan-Meier plotter. FOXM1, log-rank P = 0.00036; AURKA, log-rank P = 0.0011; CCNA2, log-rank P = 0.00018; CDKN3, log-rank P = 0.0066; MKI67, log-rank P = 0.00011; EZH2, log-rank P = 6.8e-06; CDC6, log-rank P = 3.6e-06; CDK1, log-rank P = 1.1e-05; CCNB1, log-rank P = 3.4E-05; and TOP2A, log-rank P = 0.00012. Data are presented as Log-rank P and the hazard ratio with a 95% confidence interval. Log-rank P < 0.01 was regarded as statistically significant. OS, overall survival.
Figure 8 DFS of LIHC patients overexpressed the 10 hub genes were analyzed by the GEPIA online database. Data are presented as log-rank P and the hazard ratio with a 95% confidence interval. FOXM1, log-rank P = 0.00066; AURKA, log-rank P = 0.0012; CCNA2, log-rank P = 0.0037; CDKN3, log-rank P = 0.0074; MKI67, log-rank P = 4.2e-05; EZH2, log-rank P = 1e-04; CDC6, log-rank P = 0.0044; CDK1, log-rank P = 0.00057; CCNB1, log-rank P = 2.8E-06; and TOP2A, log-rank P = 0.00053. Log-rank P < 0.01 was considered statistically significant. DFS, disease-free survival; LIHC, liver hepatocellular carcinoma.
Drug-hub gene interaction
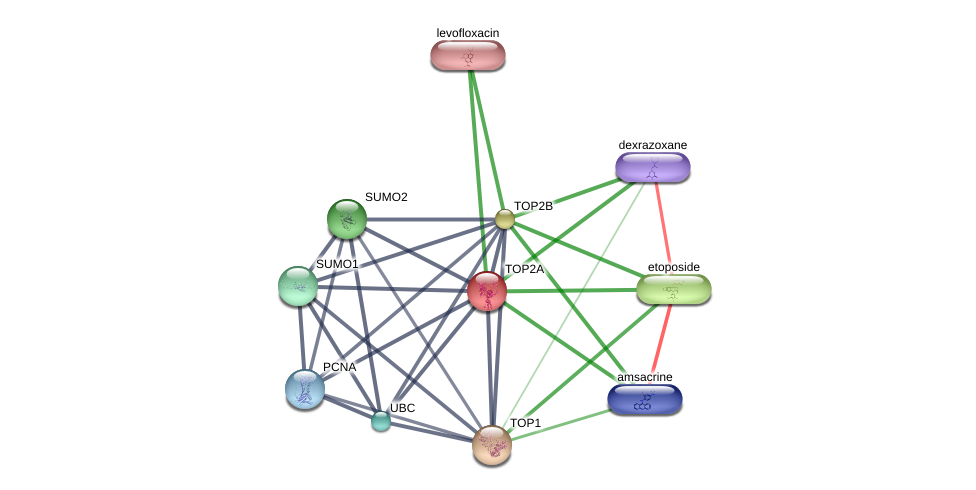
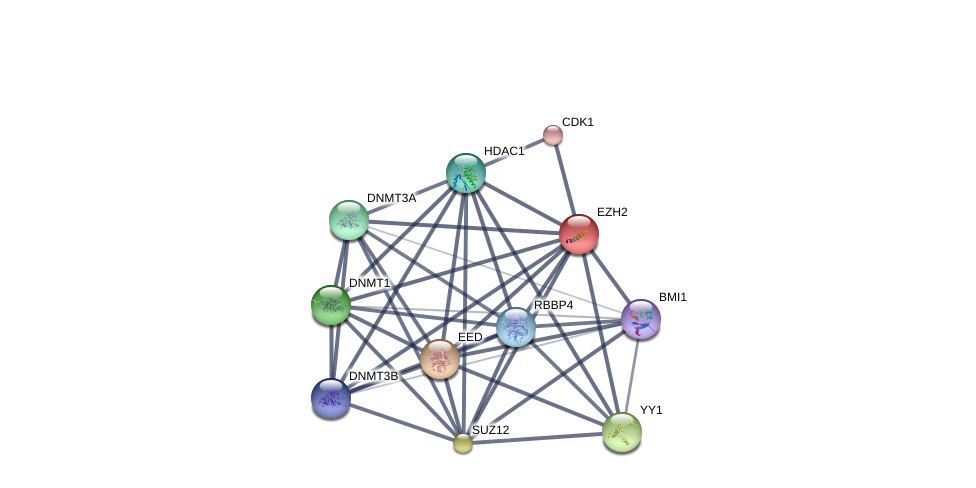
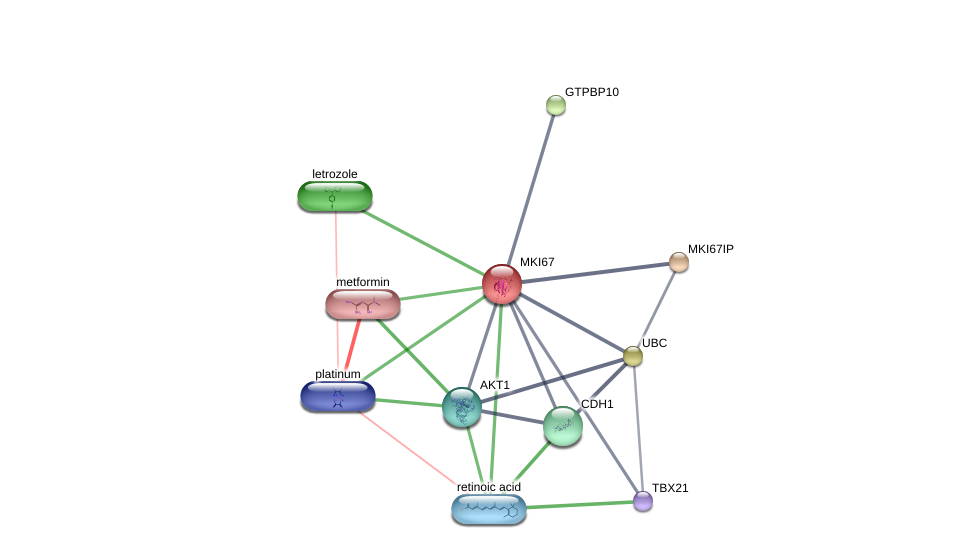
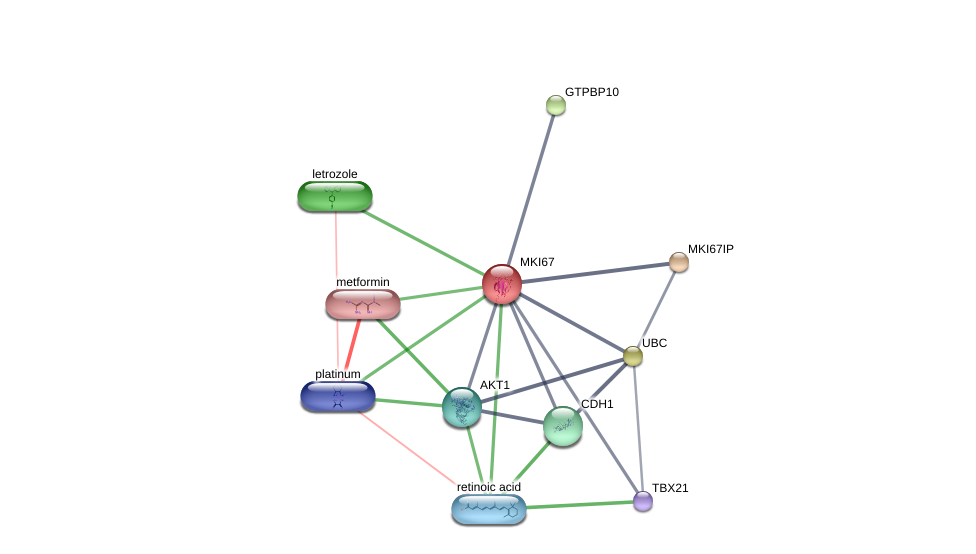
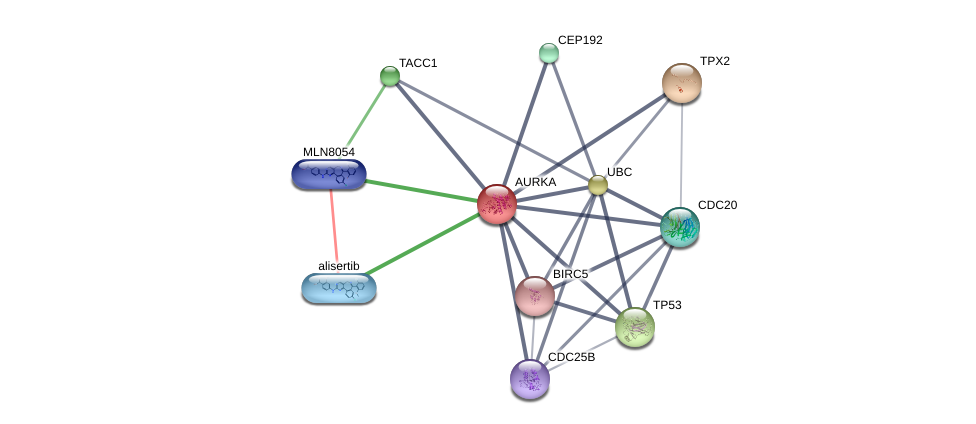
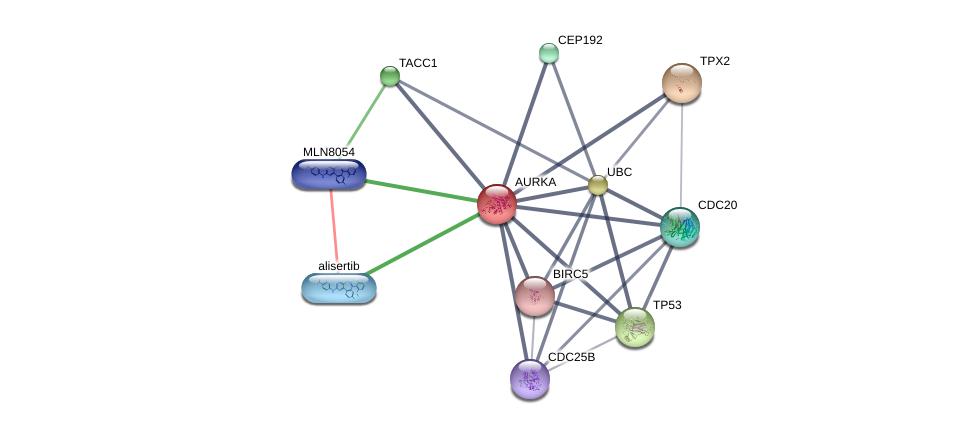
Using the DGIdb database to explore the drug-gene interactions of the 10 hub genes, 39 drugs for possibly treating HCC were matched and determined (Additional file 1:Table S1). Promising targeted genes of these drugs include AURKB, MKI67, EZH2, TOP2A. The final list only included these drugs which were approved by FDA, and several drugs have been tested in clinical trials. Paclitaxel was considered as a potential drug for cancer therapy due to its inhibition of AURKA and TOP2A. Etoposide, an inhibitor of TOP2A, could inhibit the development of cancer through inducing DNA damaging. Using STITCH database, we constructed downstream networks of AURKA, MKI67, EZH2, and TOP2A to investigate the additional effects caused by inhibitors of these genes. Our models showed that AURKA inhibition might have possible influence on TPX2, microtubule nucleation factor (TPX2), cell division cycle 20 (CDC20), tumor protein p53 (TP53), cell division cycle 25B (CDC25B), baculoviral IAP repeat containing 5 (BIRC5); MKI67 inhibition might have possible influence on AKT serine/threonine kinase 1 (AKT1), cadherin 1 (CDH1); EZH2 inhibition might have possible influence on histone deacetylase 1 (HDAC1), BMI1 proto-oncogene, polycomb ring finger (BMI1), YY1 transcription factor (YY1), DNA methyltransferase 3 alpha (DNMT3A), DNA methyltransferase 3 beta (DNMT3B), DNA methyltransferase 1(DNMT1), RB binding protein 4(RBBP4), embryonic ectoderm development(EED); TOP2A inhibition might have possible influence on DNA topoisomerase I (TOP1), DNA topoisomerase II beta (TOP2B), ubiquitin C (UBC), proliferating cell nuclear antigen (PCNA), small ubiquitin-like modifer 1 (SUMO1), and SUMO2 (Additional file 1: Figures S4-7). So far, few inhibitors of AURKA, MKI67, EZH2, and TOP2A have been tested for HCC therapy. Some of these drugs were even not regarded as anti-cancer drugs (such as levofoxacin and dexrazoxane). These data could provide new insights for targeted therapy in HCC patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}