Materials
AgNO3 as a precursor for Ag-NPs synthesis was obtained from Sigma/Aldrich, USA. Peptone, Yeast extract, Malt extract, Glucose, Starch, and Dimethyl Sulphoxide (DMSO) were purchased from Lobal Chemie, India. All materials used for cell culture experiments were obtained from Cambrex BioScience (Copenhagen, Denmark). All other chemicals and reagents were bought locally and were of analytically reagent grade from Algomhoryia Company for Chemicals, Cairo, Egypt.
Culture media and Microorganisms
Glucose Yeast Malt medium was used for actinomycetes isolation having the following composition (g/L); malt extract 10, yeast extract 3, glucose 3, agar 20, and the pH was adjusted to 7.3±0.1 at 25°C. Starch nitrate medium was used as a synthetic medium for biosynthesis of Ag-NPs (Abd-Elnaby et al. 2016) with the following composition (g/L); starch 20, K2HPO4 0.5, KNO3 1, MgSO4.7H2O 0.5, FeSO4 0.01, KCl 0.5, and pH was adjusted to 7.2±0.2 at 25°C. Nutrient agar medium was used for growth and maintenance of pathogens with the following composition (g/L); beef extract 3, peptone 5, NaCl 5, agar 20, and pH 7.
Gram-positive bacteria (Bacillus cereus ATCC 14579, Bacillus subtilis ATCC 6633, Listeria monocytogenes, Staphylococcus aureus ATCC 6538), gram-negative strains (Escherichia coli O157:H7, Klebsiella pneumonia, Aeromonas hydrophilia), and Yeast (Candida albicans), were obtained from Dar Al-Fouad Hospital at 6th October city, Cairo, and El-Mabarra Educational Hospital, Alexandria, Egypt.
Isolation of actinomycetes
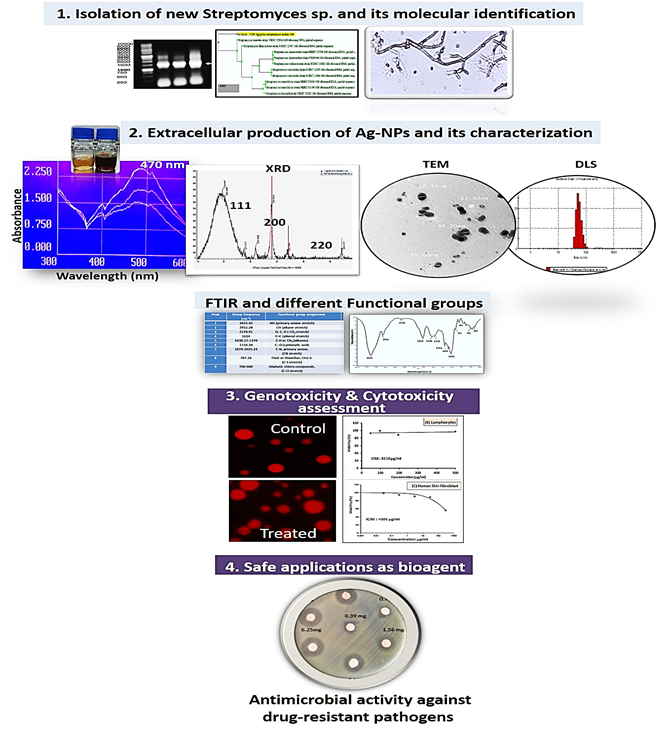
Soil samples were collected from Menuf, Minufyia governorate, Egypt in sterile plastic bags. 10 g of collected sample was suspended in 90 mL of saline solution (0.9% NaCl, w/v), serially diluted, and one mL aliquot of each dilution was plated on sterile agar plates containing Glucose Yeast Malt Agar medium and were incubated for 5 days at 30oC. Colonies which were morphologically identified as actinomycetes were selected. Pure developed single colonies were obtained by conventional streak plate technique, kept at 4°C on slants. All cultures were routinely stored in glycerol solution (25% v/v) at -80°C.
Biosynthesis of Ag-NPs
Cell free extract of the isolated actinomycete was grown in 250 mL-Erlenmeyer flask containing starch nitrate broth medium, incubated in a rotary shaker at 170 rpm and 30°C for three days. After the incubation, culture was centrifuged at 6000 rpm for 20 min, and the collected supernatant was used for the biosynthesis of Ag-NPs. 50 mL of AgNO3 solution (1 mM) were mixed with 50 mL of the actinomycete supernatant (Abd-Elnaby et al. 2016). The mixture was incubated at 30oC and 170 rpm agitation in a rotary shaker, for 24 h in dark. To study the temperature effect on the biosynthesis, the reaction mixtures were incubated at different temperature degrees (30, 45, and 60oC) for 24 h at 150 rpm in rotatory shaker. Control experiments were carried out by mixing AgNO3 solution and un-inoculated media or adding the actinomycete supernatant with double distilled H2O without AgNO3. The reduction of Ag+ ions was checked by color change of the reaction mixture from yellow to brown.
Characterization of biosynthesized Ag-NPs
UV-Visible spectroscopy (UV-Vis)
Formation of Ag-NPs during the biosynthesis was monitored based on surface-plasmon-resonance using a UV-Vis spectrophotometer (Shimadzu T80 spectrophotometer, China). One mL aliquot of biosynthesized Ag-NPs colloidal solution was scanned at wavelength ranging from 300 to 700 nm.
X-ray diffraction (XRD)
Crystalline natures of the dried biosynthesized Ag-NPs powder were investigated by X-ray diffraction (Bruker D2 Phaser diffractometer 2nd Gen.), operating at (50 kV and 0.5 mA), with a Cu anode radiation (1.54060 Å) in the angular range of 10°-70° using continuous scanning 2ϴ mode. The size of the Ag-NPs formed in the bio-reduction process was determined using Scherrer's formula (Sumadevi et al. 2021).
D = K λ/β cosθ
where K is the Scherrer constant (shape factor), λ is the X-ray wavelength (1.5418 Å), β is the width of the XRD peak at half-height, θ is the Bragg angle, and D is the grain size.
Transmission electron microscope (TEM)
The size and morphology of the biosynthesized Ag-NPs was studied by TEM (FETEM, JSM-2100F, JEOL Inc.) at Petroleum Research Institute, Cairo, Egypt. An aliquot of Ag-NPs suspension was transferred onto carbon-coated copper grid, and allowed to dry. The grid was then scanned using TEM (Phillips EM 208S) operated at 100 kV voltage.
Dynamic light scattering (DLS)
Biosynthesized Ag-NPs was analyzed using Nano-Zeta Sizer (Nano ZS, ZEN 3600, Malvern Nano, UK) to determine the distribution of particles size. DLS measures the light scattered from a laser when passes through sample that mostly depends on Rayleigh scattering from the suspended NPs and the fluctuations of the scattered light are detected at a known scattering angle θ by a fast photon detector (Fissan et al. 2014).
Fourier transform infrared (FTIR)
The biosynthesized Ag-NPs were dried then diluted (1:100) by potassium bromide. The FTIR spectrum of the sample was recorded on a FTIR spectrometer 8000 series, with KBr in the wavenumber region of 4,000-400 cm-1 at a resolution of 4 cm-1. To identify the functional groups found in the tested sample, the spectral data recorded were compared with the reference data chart.
Molecular identification and phylogenetic analysis
The selected actinomycetes isolate was identified using 16S rRNA gene partial sequencing method (Kim et al. 2011). Genomic DNA of the selected actinomycetes isolate was extracted using genomic DNA extraction Kit (Intron, Biotechnology, Korea). PCR amplification of 16S ribosomal DNA (16S rDNA) was performed with a set of universal primers; 27F-Forward primer 5′AGA GTT TGA TCC TGG CTC AG 3′ (20 mer) and 1492R-Reverse primer 5′CTA CGG CTA CCT TGT TAC GA 3′ (20 mer). The PCR amplification was performed using programmed thermal cycler (Thermo Fisher Scientific, USA). The produced amplicons of the selected isolate (PCR product) was tested for its quality by electrophoresis in Bio-Rad submarine (8x12 cm) using agarose gel (1%) and compared with 1kb DNA ladder (Intron Biotechnology, Korea). DNA banding patterns of 16S gene amplicons were visualized using a UV-transilluminator (Thermo Fisher scientific, USA) under UV light. PCR products were purified by using gene JET™ genomic DNA purification kit (Intron Biotechnology, Korea) according to the manufacturer’s instructions. Then sequenced using forward and reverse primers with ABI 3730xl DNA sequencer. Nucleotide bases obtained after sequencing were identified and compared with similar sequences retrieved from the GenBank database within the National Center for Biotechnology information (NCBI) (http://www.ncbi.nlm.nih.gov/GenBank/index.html) using nucleotide Basic Local Alignment Search Tool (BLAST) Gene Sequences in the database website (http://www.ncbi. nlm.gov/BLAST/). The nucleotide sequences of the 16S rRNA genes was deposited under accession number MT071505, and the phylogenetic analysis of sequences was created using Alignment Search Tools (BLAST).
Qualitative and quantitative assessment of antimicrobial activity
Bacterial strains and growth conditions
The antimicrobial activity of the biosynthesized Ag-NPs was tested against different Gram-positive bacteria (L. monocytogenes, S. aureus, B. subtilis, B. cereus,), Gram-negative bacteria (E. coli O157:H7, K. pneumonia, A. hydrophilia), and yeast (C. albicans). All bacterial strains were grown on nutrient broth, except L. monocytogenes was grown on brain heart infusion (Oxoid, ltd, England) and the yeast was grown on Wickerham medium. Stock inoculum suspensions of the pathogenic strains were freshly prepared by picking colonies from 24 h cultures grown on at 37⁰C and suspended in sterile saline solution (0.9% NaCl, w/v). The optical density of pathogens was adjusted to achieve turbidity equivalent to 0.5 McFarland standard, approximately (1×108 CFU/mL) for bacteria, and (1x106) for C. albicans (Andrews 2001).
Disc diffusion method
Disc diffusion method is the common method used as a preliminary screening test prior to quantitative minimal inhibitory concentration (MIC) determination. Microbial inocula were spread on plates containing appropriate medium using sterile cotton swab, and plates were allowed to dry for 15 min at room temperature. Sterile filter paper discs (Hi media) were saturated by 40 μL of two-fold serially diluted concentrations of biosynthesized Ag-NPs in the range (0.39-25 mg/mL). Discs saturated with the actinomycete supernatant and AgNO3 were used as a negative and positive control, respectively. The plates were incubated at 4oC for 1h to allow diffusion of Ag-NPs into agar medium, and were incubated at 37⁰C for 24h (Wikler 2006). After incubation, inhibition zones diameter was measured in mm, and MIC was determined as the lowest concentration of biosynthesized Ag-NPs that produced an inhibition zone after 24h of incubation.
Agar dilution method
Brain Heart Infusion (BHI) and nutrient agar (NA) media were used for the growth of L. monocytogenes and K. pneumonia, respectively. Two-fold serial dilutions of biosynthesized Ag-NPs were prepared in molten BHI agar and NA medium, cooled down to 45°C with desired final concentrations. Using pour-plate method (Klančnik et al. 2010), 100 μL of L. monocytogenes or K. pneumonia bacterial suspensions (0.5 Mcfarland~1:2×108 CFU/mL) were inoculated into sterilized agar petri-plates containing appropriate medium, and incubated at 37°C for 24-48 h. An inoculated agar plate without Ag-NPs was served as a positive control and another one without inoculum as a negative control.
Broth macro-dilution method, MIC, and MBC determination
Broth macro-dilution was done according to Andrews (2001) method. The biosynthesized Ag-NPs were added to 10 mL of BHI broth medium and nutrient broth to give final concentrations with respect to the results obtained by the agar dilution method. 100 μL from the diluted bacterial culture of L. monocytogenes or K. pneumonia (0.5~Mcfarland 1:2x108 CFU/mL) were inoculated in growth media containing the desired concentration of Ag-NPs, well shaken, and incubated at 37⁰C for 24h. Bacterial growth was followed by plating 1 mL from the incubated culture tubes and control on suitable media after serial dilutions. After 24h of incubation, plates were compared with positive control and the bacterial colony number was calculated as (CFU/mL). The MIC is the lowest concentration of biosynthesized Ag-NPs resulting in significant reduction (99%) of pathogen viability after 24h of incubation. The concentration where 100% of microbial growth was inhibited as compared to the negative control (media only) was designed as MBC value. The tolerance level of pathogen towards the biosynthesized Ag-NPs was calculated using the following formula (May et al. 1998):
Level of Tolerance = MBC/MIC
Scanning electron microscope (SEM) imaging
The effect of biosynthesized Ag-NPs on pathogens was studied using SEM, micrographs were captured in a Jeol JSM- 5300 SEM operated between 15 and 20 KeV at Faculty of Science, Alexandria University, Alexandria, Egypt. Samples were prepared according to Tamboli and Lee (2013) method with some modifications as follows; 10 mL of fresh bacterial culture of L. monocytogenes and K. pneumonia grown in Luria-Bertani medium for 18h were treated with biosynthesized Ag-NPs at the detected MIC, then incubated for 3 and 6h. Treated bacterial cells were collected by centrifuging at 6000 rpm for 10 min. Culture supernatants were discarded and the treated cells were washed by phosphate saline buffer (pH 7.4) to remove excess of Ag-NPs precipitated on the cells, then centrifuged to collect washed cells. Samples were fixed by immersing immediately in equal volume of a fixative solution (1% glutaraldehyde, 4% paraformaldehyde in 0.1M sodium-cacodylate) at 4⁰C for 24 h. Specimens were then post fixed in 2% OsO4 in the phosphate saline buffer (pH 7.4) at 4⁰C for 2 h. Samples were washed in the buffer and dehydrated at 4⁰C through graded series of ethanol, and dried by N2 gas. Finally, the samples were mounted using carbon paste on AL-stub using double-sided conductive tapes, and coated with gold up to a thickness of 400 A in a sputter-coating unit (JFC-1100 E) provided at the SEM lab.
Cytotoxic assessment of biosynthesized Ag-NPs
Cell viability and cytotoxicity of biosynthesized Ag-NPs was tested on normal Human Skin Fibroblast (HSF) cell-lines using sulforhodamine B protein (SRB) assay, and on Peripheral Blood Lymphocytes normal cells, as well as Human Hepatocellular Carcinoma (HepG-2) cell-lines using MTT assay. Normal HSF cell-lines were obtained from Nawah Scientific Inc., (Mokatam, Cairo, Egypt). Cells were maintained in Dulbecco’s Minimum Essential Medium (DMEM) supplemented with 100 mg/mL of streptomycin, 100 U/mL of penicillin and 10% of heat-inactivated fetal bovine serum in humidified CO2 (5% v/v) at 37°C. Aliquots of 100 μL cell suspension (5x103 cells) were distributed in 96-well plates and incubated for 24h. Cells were treated with aliquot of 100 μL media containing Ag-NPs (0-300 μg/mL) concentrations. After 72h of exposure, cells were fixed using 150 μL of TCA (10%), and incubated for 1h at 4°C. The TCA solution was removed, and cells were washed 5-times using double distilled water. Aliquots of 70 μL SRB solution (0.4% w/v) were added and incubated at 25⁰C in darkness for 10 min. Plates were washed 3 times with acetic acid (1% v/v) and allowed to air-dry. To dissolve the protein-bound SRB stain, 150 μL of TRIS (10 mM) was added, and the absorbance was measured at 540 nm using a BMGLABTECH®-FLUO star Omega microplate reader, Ortenberg, Germany (Skehan et al. 1990).
Hep-G2 cell-lines (ATCC, USA) were used to evaluate the cytotoxic effect of the biosynthesized Ag-NPs using 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were routinely cultured in DMEM, which is supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL streptomycin-sulphate, 250 ng/mL amphotericin B, and 100 U/mL sodium penicillin-G. All cells were maintained in humidified air containing 5% CO2 at 37°C. For sub-culturing, the monolayer cells were harvested after trypsin/EDTA treatment at 37°C. Peripheral Blood Lymphocytes were isolated from fresh blood samples according to Kizhakeyil et al. (2019). Lymphocyte cell suspension was centrifuged at 2000 rpm for 10 min, washed twice then suspended in incomplete RPMI-1640. Lymphocyte cell viability was checked using Trypan blue stain (Sigma) and was above 90%. The cells (2.5X105 cells/mL) were cultured in RPMI-1640 supplemented with 10% FBS as triplicates of 200 µL/well into flat-bottom 96-well tissue culture plates (Griener), and incubated with serial concentrations of biosynthesized Ag-NPs (48-500 µg/mL concentration) for 48h in a humidified 5% CO2 atmosphere. HepG-2 cells (0.5X105 cells/well), in serum-free medium, were plated in a flat bottom 96-well microplate, and treated with 20 µL of different Ag-NPs concentrations in the range (62.5-500 µg/mL) for 24h at 37ºC, in a humidified 5% CO2. After incubation, 40 µL MTT solution/well were added after the removal of media and incubated for an additional 4h. MTT crystals were solubilized by the addition of 180 µL of DMSO/well, then plates were shacked at room temperature, followed by photometric determination of the absorbance at 570 nm using microplate ELISA reader (SunriseTM, Tecan Group Ltd. Männedorf/Switzerland). Each concentration was carried out in triplicate, the average and standard deviation were calculated. Data were expressed as the percentage (%) of relative viability compared with untreated cells, with cytotoxicity indicated by <100% relative viability. The extent of MTT reduction was quantified, where the number of viable cells was directly proportional to the intensity of formazan dark-blue color (Hansen et al. 1989). IC50 was calculated from dose response curve and the percentage (%) of relative viability was calculated using the following equation:
[Absorbance of treated cells/Absorbance of control cells)] X 100
In vitro genotoxicity alkaline comet assay
The comet assay was assessed according to Singh et al. (1988) with some modifications. Treated cells were incubated with different concentrations of biosynthesized Ag-NPs for 12h, under proper sterilized anaerobic conditions. Cells were washed by phosphate saline buffer (pH 7.4), resuspended in 0.75-1% low dissolving agarose (Bio-Rad) and were spread onto slides covered by 1% typical agarose. Slides were left to solidify at 40⁰C for 5 min, then were placed in lysis buffer (2.5M NaCl, 10 mM Tris, 100 mM EDTA, 1% TritonX100, 10% DMSO, pH 10) overnight at 40ºC. Slides were placed in unwinding buffer (300 mM NaOH, 1 mM EDTA, pH ˃13) for 40 min at 40ºC. Electrophoresis was carried out for 20 min at 25V (1.0 V/cm), and 300 mA. Finally, the slides were neutralized using 0.4M Tris buffer (pH 7.5), and were stained with ethidium bromide (20 µg/mL), then examined using a fluorescence light microscope (Leica DMi8 S-platform, Germany), 100×objective lens. The tail intensity %, head intensity, and tail moment were assessed using Comet examine IV software.
Statistical analysis
All experiments were conducted in triplicates, the mean and all standard deviations were calculated using Excel software (Microsoft office, 2010). Data were expressed in their mean values ± SD (standard deviation).
{kind=link}