Quality Control of scATAC-seq
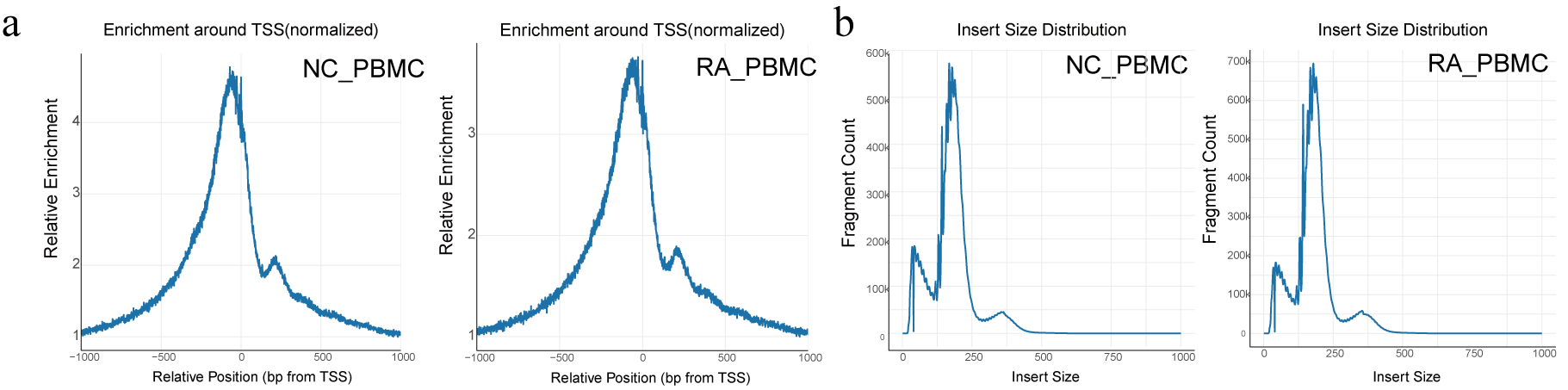
RA group and NC group were acquired and analyzed by scATAC-seq as the workflow shown. After quality control, 16,407 cells and 5,344 unique fragments were retained. At single-cell resolution, RA_PBMC enriched TSS fragments four times in the proximal region relative to the distal region, and NC_PBMC enriched TSS fragments five times, which reflected that the proportion of fragments captured in the open chromatin was higher than that in the closed chromatin. Besides, we noted distribution characteristics of nucleosome fragment between the RA group and NC group. The recovered library from RA_PBMC was sequenced to an average depth of 22,743 raw reads per cell, generating chromatin accessibility profiles for 8014 cells with a median of 5,345 unique fragments per cell. Simultaneously, the library of NC_PBMC generates profiles of 8,393 cells with 23,035 original reads per cell and 5,344 unique fragments per cell.
Identification of primary cell types among PBMCs
The RA group and NC group were acquired and analyzed by scATAC-seq as shown in the workflow (Figure 1a). Quality control of the scATAC-seq profile was showed in figure 1b and figure 1c. In detail, we observed the signal distribution map around TTS after normalization of NC_PBMCs and RA_PBMCs library and the length distribution of corresponding inserts for each sample. Here, we used the activity of known cell marker genes to identify and annotate 9 clusters (Figure 1b), including T lymphocytes (T cells; clusters 0, 1, 3, and 5), natural killer cells (NK cells; cluster 2), monocytes (cluster 4), B lymphocytes (B cells; cluster 6) and dendritic cells (DCs; cluster 8). More specifically, T cells can be identified by CD3D, CD3G, LEF1, CD8A and IL2RA[10, 11]; NK cells by GZMB, NKG7, and KLRD1[10, 12]; monocytes by CD68, CD14, and ITGAM[10, 13]; B cells by CD79A, CD19 and MS4A1[10, 12]; and DCs by IL3RA[10]. Select marker genes and the corresponding expression quantities are shown in Figure 1c.
In addition, scATAC-seq enables cell type identification by cell type-specific transcription factor motifs[14]. For the fragments that overlapped the list of TF motifs, annotations of cell-type specificity were created for the most significantly enriched TF motifs (P-value < 0.01, fold change value, FC>1.5) in each cluster with no apparent difference between the RA_PBMC and NC_PBMC groups to serve as an alternative identifying feature for each cluster. Notably, more than 20 TF motifs in B cells were identified. Thus, we selected the top 10 TF motifs: POU2F3, IRF1, POU5F1B, STAT1:: STAT2, POU2F, POU1F1, POU2F2, POU3F3, POU5F, and POU3F1 (Figure 2a). In addition, we found 13 TF motifs in NK cells, including EOMES, TBX2, TBR1, TBX20, and TBX21 (Figure 2a). Furthermore, we found 95 TF motifs in monocytes. The top 10 TF motifs in monocytes were FOSL2::JUN, FOSL1::JUN, FOS, FOSL1, FOSL1::JUND, FOSL1::JUNB, JUN(var.2), JUNB, FOS::JUND, and JUND (Figure 2a), while there were 20 TF motifs including GATA3, ELF2, ELK1, CEBPB, ELK3, GATA5, FEV, CEBPE, CEBPA, ELF5 in DCs. Interestingly, 7 TF motifs were identical in DCs and monocytes: CEBPB, CEBPE, CEBPA, ELF5, NFIL3, CEBPG, and HLF (Figure 2b), because some DCs differentiate from monocytes. For T cells, there was no TF motif with an FC value higher than 1.5. In summary, we concluded the identified markers genes and type-specific transcription factor motifs in the scATAC-seq experiments (Table 2).
Differential chromosome accessibility analysis in rheumatoid arthritis
The distribution of immune cells and their regulatory functions are variously related to the occurrence of autoimmune diseases. Comparing the ratios of immune cells in the PMBC_RA with PBMC_NC groups (Table 3, Figure 2c), a significant difference was observed between B cells and T cells (Chi-square test, P-value < 0.01, FDR<0.05). Notably, the ratio of T cells in the PBMC_RA group was lower than that in the PBMC_NC group, implying a possible mechanism by which fewer T cells curb inflammation in the RA environment, a finding that is consistent with the hypothesis suggesting that regulatory CD8+ T cells play a role in preventing the development of autoimmune diseases[15]. In contrast, there were more B cells in the RA group than in the NC group (P-value < 0.01, FDR<0.05), suggesting that B cells in peripheral blood can promote RA pathogenesis by changing the number of cells. When calculating the different peaks (P-value < 0.05, FC>1.2) in the PBMC_NC and PBMC_RA groups, we found no different peaks in T cells, 51 in NK cells, 11 in monocytes, 149 in B cells, and 665 in DCs (Figure 2d). Moreover, the differential accessibility of TF motifs (P-value < 0.05, FC>1.2) for each immune cell type was selected. Consequently, we identified 56 TF motifs in RA, including three motifs (CEBPG (var. 2), NFE2L1, MAF:: NFE2) in monocytes, six motifs (NRF1, PHOX2A, TCFL5, ZBTB14, PHOX2B, PROP1) in B cells, one motif (TCFL5) in T cells and 46 motifs in NK cells (Figure 2e). However, no differential TF motifs were observed in DCs, indicating that the transcriptome signatures of these cells did not change in the RA environment.
We identified a total of 22 subclusters among B cells, DCs, monocytes, NK cells, and T cells (Figure 3a) with further cluster analysis. The results show that the cell number ratios of subcluster 0 B cells (B-0), subcluster 3 NK cells (NK-3), and subcluster 3 T cells (T-3) in the PBMC_RA group were more eight-fold greater than those in the PBMC_NC group. In addition, the cell number ratios in B-2, monocyte-1, NK-1, and T-5 in the PBMC_RA group were also higher than in the PBMC_NC group. However, the cell number ratio of subcluster 3 B cells (B-3), subcluster three monocyte cells (monocyte-3), subcluster 0 natural killer cells (NK-0), subcluster four natural killer cells (NK-4), subcluster 0 T cells (T-0), subcluster 4 T cells (T-4) and subcluster 6 T cells (T-6) in the PBMC_RA group was smaller than that in the PBMC_NC group, especially for NK-0 and NK-4, in which it was decreased by more than three-fold. Furthermore, no obvious difference in the number of cells was observed in the remaining subclusters in the PBMC_RA and PBMC_NC groups. We profiled a figure for the cell ratio, as mentioned above (Figure 3b). Furthermore, the differential accessibility of TF motifs (P-value < 0.05, FC>1.2) in each subgroup of PBMCs is shown in Figure 3c.
Functional analysis of significantly enriched peaks in the PBMC_RA group
According to the epigenomic analysis, we selected peaks with an obvious fold change value higher than 1.2 for the GO and KEGG analysis. Based on the KEGG analysis, NKs participate and mediate RA progression through virus infection-related pathways, such as human CMV, human immunodeficiency virus 1, Kaposi sarcoma-associated herpesvirus, Epstein-Barr virus, and human T-cell leukemia virus (Figure 3d). In addition, NKs were also active in Th17 cell differentiation. This result was related to another study showing that helps T (Th) cells participate in RA pathogenesis, especially Th17 cells, which produce IL-21, IL-22, and TNFα[16]. Similarly, we found that DCs are regulated through human cytomegalovirus (CMV) infection (Figure 3e), a finding consistent with research showing that RA patients usually have CMV[17].
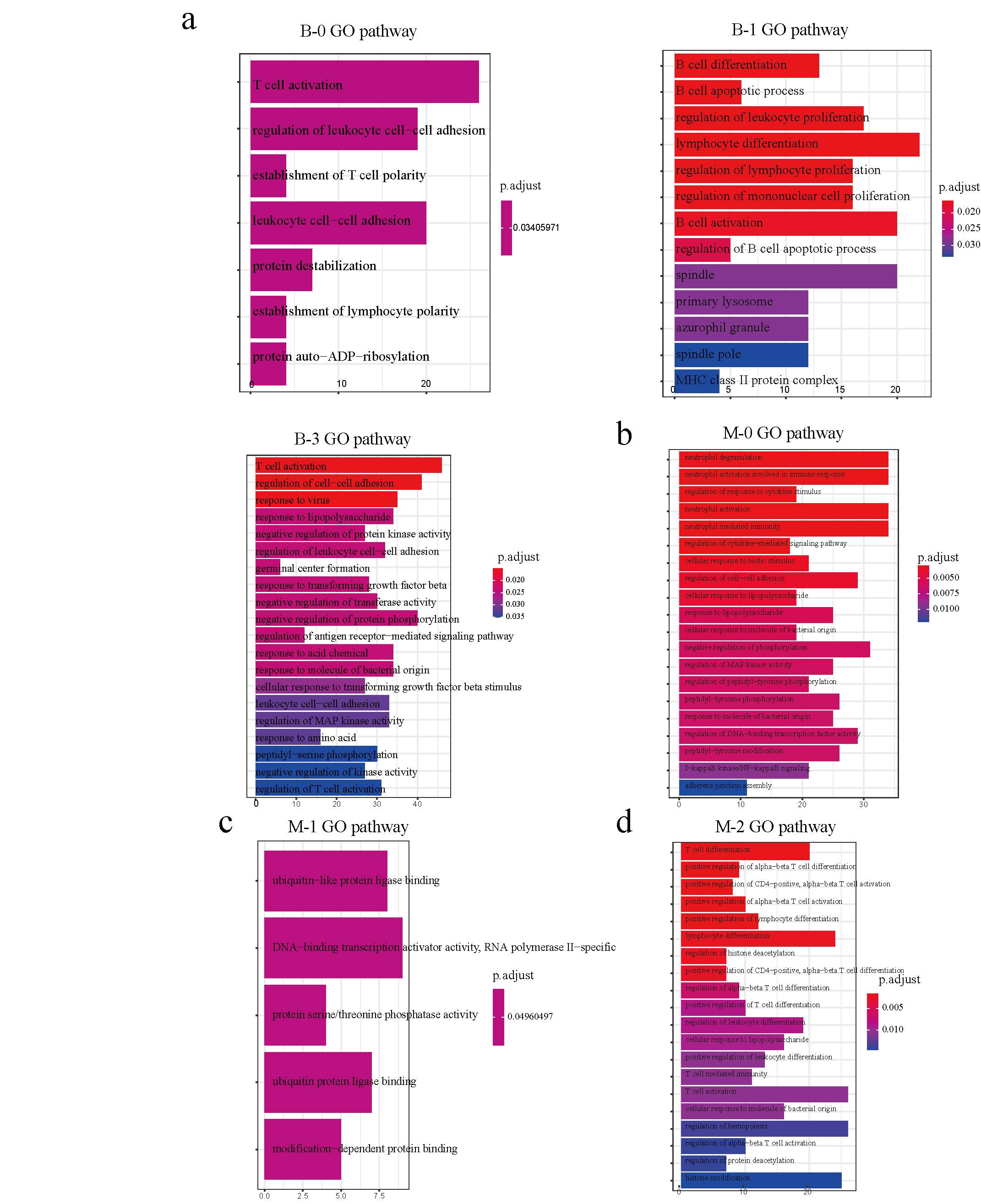
Similarly, after further clustering of five major types of PBMCs, we found cell-type-specific functions through GO enrichments considering peak-related genes in subclusters, except for the B-2 and T-1 subclusters. Here, the B-0 and B-3 subcluster genes were found to play roles in T cell activation. At the same time, B-1 genes are involved in B cell differentiation and activation. In addition to T cell activation, B-3 genes also participate in regulating cell−cell adhesion, negative regulation of protein phosphorylation, regulation of MAP kinase activity, and T cell activation regulation. In monocytes, M-0 genes are involved in the negative regulation of phosphorylation, regulation of MAP kinase activity, neutrophil degranulation, activation, and mediation of immunity. M-1 genes are regulated through DNA-binding transcription activator activity; RNA polymerase II-specific. M-2 genes are active in histone modification, T cell activation, and differentiation.
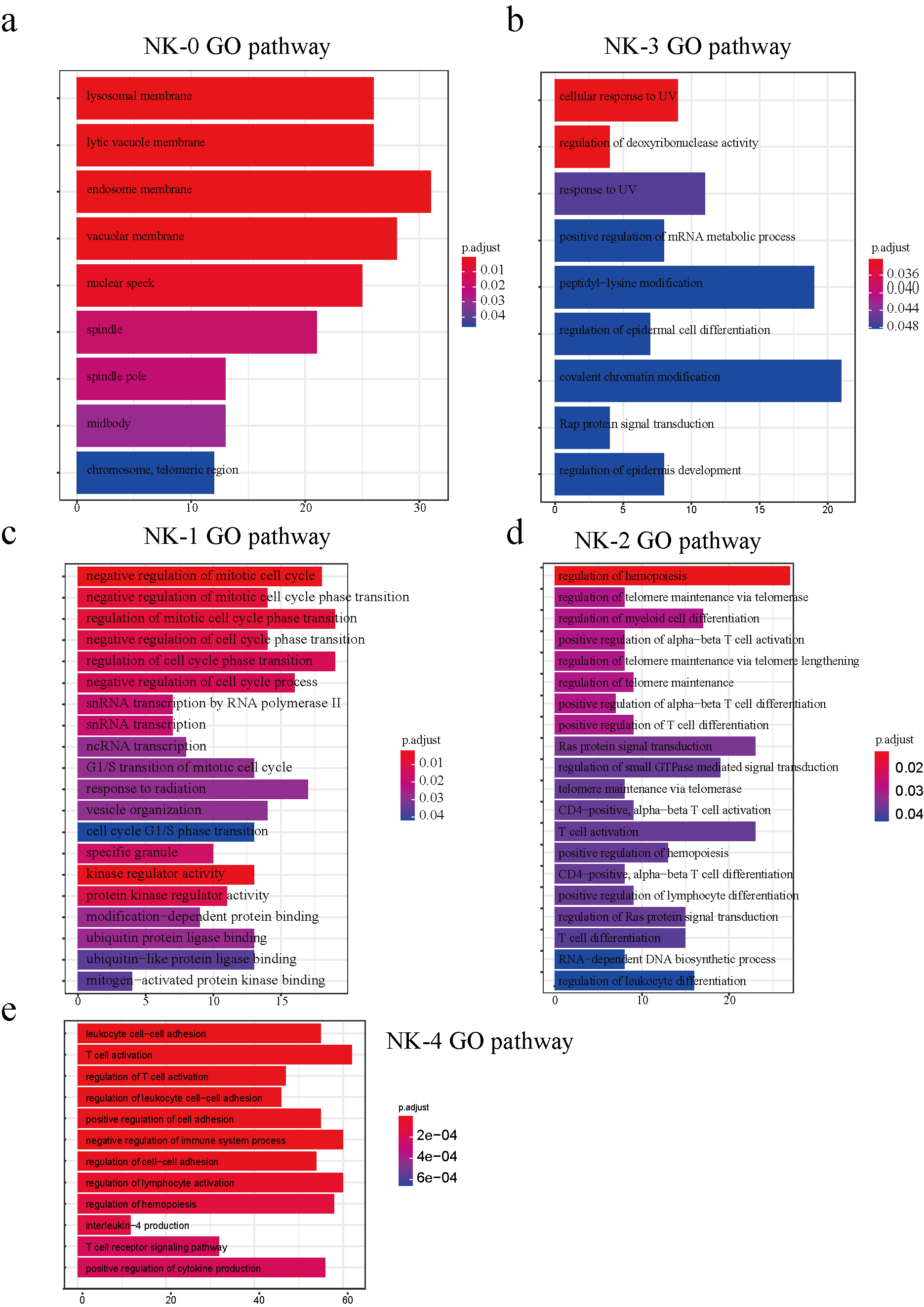
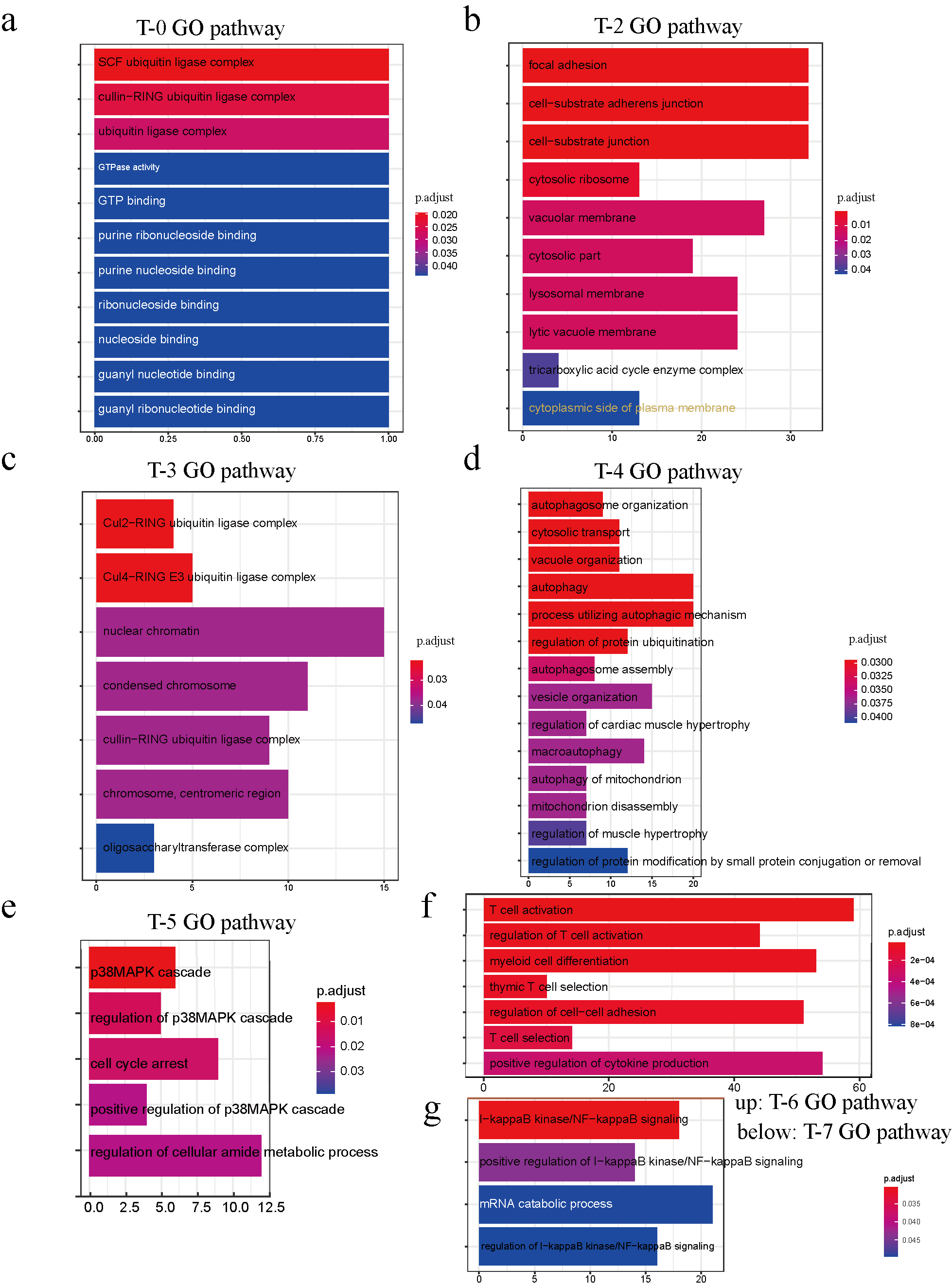
Similarly, the M-3 subcluster genes participate in dephosphorylation, regulation of GTPase activity, regulation of small GTPase-mediated signal transduction, positive regulation of cell adhesion, and T cell activation. In NK cells, peak-related genes in the NK-0 subcluster are mainly involved in the endosome membrane; NK-1 genes participate in the regulation of mitotic cell cycle phase transition and kinase regulator activity; NK-2 genes take part in Ras protein signal transduction, regulation of small GTPase-mediated signal transduction, and T cell activation; NK-3 genes have effects on the cellular response to UV; NK-4 genes are mediated through T cell activation, regulation of lymphocyte activation, positive regulation of cytokine production and regulation of GTPase activity. With regard to subtypes of T cells, genes are abundant in the ubiquitin ligase complex in the T-0 subcluster, focal adhesion in T-2, and the Cul4−RING E3 ubiquitin ligase complex in T-3. In addition, considering biological process, the T-2 subcluster genes are active in cell−substrate adherens junction; T-4 in autophagy; T-5 in the regulation of cellular amide metabolic process and p38 MAPK cascade; T-6 in T cell activation and positive regulation of cytokine production; and T-7 in I−kappaB kinase/NF−kappaB signaling (supplemental figure 4g).
Specifically, B-3 and M-0 genes are active in the same pathway of regulation as MAP kinase activity (Figure 4a and Figure 4b), which is enriched by 33 genes in the B-3 subcluster and 25 genes in the M-0 subcluster. Among these genes, five genes are in both the B-3 and M-0 subclusters: MAP3K3, SPAG9, PTPRC, PTPN1, and SRC.
The seminal discovery of TFs in the PBMC_RA group
A total of 434 significantly TF-enriched TF motifs were identified in the RA_PBMC group (P-value < 0.05, FC>1.2) through further cell clustering based on the scATAC-seq analysis. We discovered two (PTPRC and SPAG9) of the 5 intersecting genes with differential accessible TF-binding sites in B-3 and M-0 subclusters of RA patients compared to healthy controls (P-value < 0.05, FC>1.2). In addition, PTPRC related to RA was identified from Gene Data Mining to Disease Genome Sequence Analysis (www.genecards.org), and the relevance scores were higher than 9. More specifically, 71 TFs could be involved in regulating two genes (PTPRC and SPAG9), and only 10 TFs had highly accessible binding sites (GATA6, IRF2, ZNF384, HNF1B, DMRTA2, MEF2A, NFE2L1, CREB3L4 (var. 2), FOSL2::JUNB (var. 2), and MEF2B). Therefore, 2 TFs (GATA6 and IRF2) in B-3 and the 8 TFs in M-0 (ZNF384, HNF1B, DMRTA2, MEF2A, NFE2L1, CREB3L4 (var. 2), FOSL2::JUNB (var. 2), and MEF2B) contributed for RA pathogenesis by regulating corresponding target genes and MAP kinase activity (Figure 4c and 4d). To figure out the cell traits of these subcluster, we further identified the enriched motifs in these subcategories in PBMC_(p<0.05, FC>1.2) (Table 4).
To validate the expression of two genes in monocytes (PTPRC, SPAG9), we took the peripheral blood of another three RA patients and healthy donors. Using these samples, monocytes were isolated and used for extracting their RNA for qPCR experiments. Compared to healthy controls, differential expression of two genes (SPAG9 and PTPRC) was observed in RA patients (SPAG9, 1.673 ± 0.8945 vs. 1.013 ± 0.1673, p = 0.0045; PTPRC, 0.5878 ± 0.3841 vs. 1.007 ± 0.1203, p =0.0066) (Figure 4e).
{kind=link}
{kind=link}
{kind=link}
{kind=link}