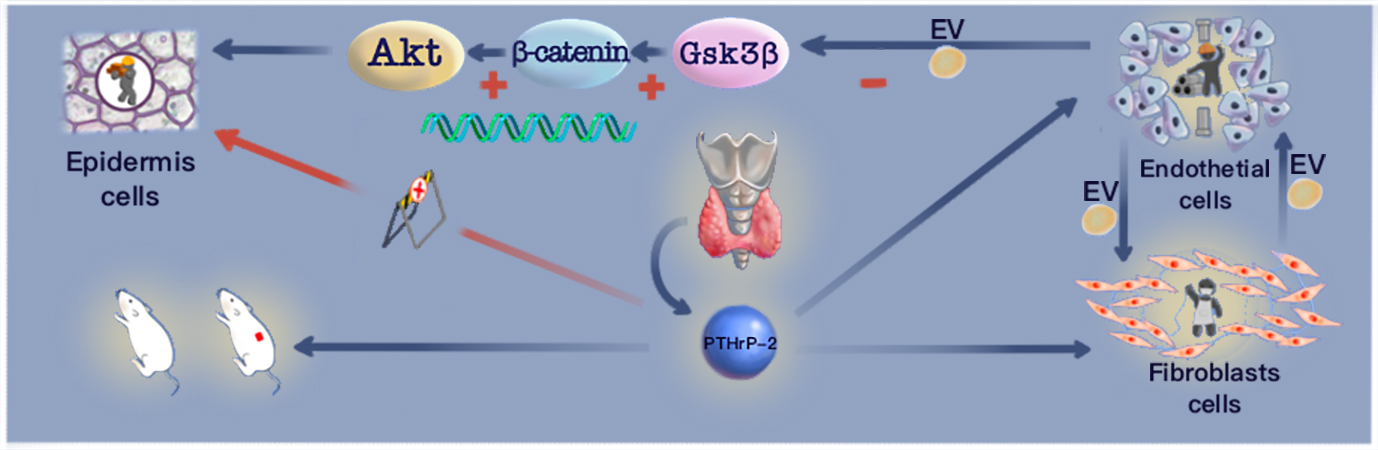
Synthesis and loading of PTHrP-2
To synthesize PTHrP-2, the FMOC/tBu solid-phase method was used(27). The whole sequence of PTHrP-2 is S [PO4] VSEI-QLMHN-LGKHL-NSMER-VEWLR-
KKLQD-VHNF-EEE. The three glutamic acid (Glu) residues at the C terminal and the phosphorylated serine (Ser) residue at the N-terminal are the major variations in PTHrP-2 relative to the 1-34 amino acids of PTH. Gel filtration was applied for initial purification of the crude peptide. In order to release PTHrP-2 sustainably in vivo, PTHrP-2 loading was applied in calcium alginate hydrogel. According to previous studies(28, 29), 2% sodium alginate solution was mixed with PTHrP-2, and 1.5% CaCl2 solution was mixed into gel. The final product was PTHrP-2@Ca-Alg.
In vivo study
Induction of diabetes and excisional wound splinting model preparation
Forty-five male Sprague-Dawley rats aged 8 weeks were selected to construct a diabetic rat model according to a previously established method. All protocols obtained approval from the Animal Care and Experimental Committee of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital. Before operation, the rats fasted for one night to measure the baseline blood glucose levels. Streptozotocin (65 mg/kg b.w., i.p.) was intraperitoneally injected, and blood glucose levels were measured at three time points, on days 1, 3 and 7. After observation for two weeks, 24 rats with glucose levels of over 300 mg/dl were selected as experimental rats for follow-up operation. According to the results of the in vitro experiment, the diabetic rats were divided into three groups, control, Ca-Alg, and PTHrP-2@ Ca-Alg, to evaluate the ability of PTHrP-2@ Ca-Alg to repair diabetic skin wounds.
Animals and surgical procedure
A rodent model of full-thickness skin wounds in diabetes was established. After the wound was successfully established, Ca-Alg and PTHrP-2@ Ca-Alg were placed on the wound surfaces of the animals. After surgery, sterile gauze was used to fix the wound surface. The rats were observed every day to ensure that the dressings were intact. After the operations, the animals were placed in a controlled temperature environment and continued to be fed with the diabetic diet, and their bedding was replaced every day.
Measurement of wound size reduction
Postoperative photos were taken with a camera (Canon, Japan) at the following four time points: day 0, day 3, day 7 and day 14. A model diagram of wound repair was constructed, and the changes in wound area and repair status were analyzed by ImageJ. The amount of wound closure was determined using the formula percent wound size reduction = ¼ [(A0-At)/A0] 100, where A0 was the initial wound area (t ¼ 0), and At was the wound area at each time point.
Microfil perfusion and microcomputed tomography
Microfil was used to evaluate neovascularization during wound healing in the diabetic rats. The experimental rats were euthanized 14 days after surgery. The hair was removed from the chest of each rat, and scissors were used to cut open the chest. After clamping the descending aorta and incising the inferior vena cava, the left ventricle was penetrated with an angiocatheter. Then, 100 ml of heparinized saline and 20 ml of Microfil (MV-122; Flow Tech, USA) were successively perfused at 2 ml/min. To ensure the polymerization and solidification of the contrast agent, the experimental samples were incubated at 4°C overnight. On the second day after the operation, the sample was pruned and scanned with microcomputed tomography (Micro CT) at a resolution of 9 mm to detect new blood vessels. Using 3D Creator software, 3D images were reconstructed. The blood vessel area and number of blood vessels in the wound were also determined using this software.
Histologic, immunohistochemical and immunofluorescence analysis
For histology, the samples were dehydrated, embedded in paraffin and sliced into sections (~6μm thick). Neuroepithelial length and collagen deposition were observed via hematoxylin and eosin (H&E) and Masson’s trichrome staining. Immunohistochemistry and immunofluorescence were applied to observe angiogenesis and fibroblasts in the wound field. For immunohistochemistry, the sections were rehydrated and treated with antigen retrieval. After incubation with the primary antibody against CD31 (1:200, Abcam, Cambridge, UK) at 4°C overnight, the sections were incubated with a biotinylated secondary antibody and an ABC complex and stained with DAB substrate. All sections were counterstained with hematoxylin and observed under a light microscope. For immunofluorescence, the sections were rehydrated and blocked with 1.5% goat serum (Merck-Millipore). After incubation with primary antibodies against CD31 (1:200, Abcam, Cambridge, UK) and α-SAM (1:50, Abcam, Cambridge, UK) at 4°C overnight, the sections were incubated with Alexa Fluor 488- and Cy3-conjugated secondary antibodies and DAPI (Sigma-Aldrich) for visualization. The sections were observed via confocal laser scanning microscopy. Angiogenesis was determined in six sections from different samples. For each section, six high-power fields containing the entire portion of the wounds were randomly observed, and the newly formed blood vessels were evaluated. All counting procedures were conducted separately by two pathologists.
PTHrP-2 effects on endothelial cells, fibroblasts and epithelial cells
Cell culture
HUVECs (Sciencell Research Laboratories, San Diego, CA, USA) were cultured in complete endothelial cell medium (ECM, Sciencell, USA) containing 2.5% fetal bovine serum (FBS, Sciencell), 1% endothelial cell growth supplement (ECGS, Sciencell) and 1% penicillin-streptomycin (P/S, Sciencell). Only the HUVECs from early passages (passages 2~7) were used in the subsequent experiments. HFF-1 cells (SCSP-109, Stem Cell Bank, Chinese Academy of Sciences) and human immortalized epidermal cells (HaCaTs) (AD4013, ATCC) were cultured under humidified conditions in serum-free Dulbecco's modified Eagle's medium (DMEM; GIBCO; Invitrogen Pty Ltd., Australia) supplemented with 2.5% FBS (Sciencell) and 1% P/S (Sciencell). Cells were cultured in a humidified 37°C/5% CO2 incubator.
Cell proliferation and migration
The proliferation of HUVECs, HFF-1 cells, and HaCaTs was analyzed with the CCK-8 method. HUVECs were cultured in medium under control conditions or with 0.1 nM, 1 nM or 10 nM PTHrP-2 (n = 4). The cells were inoculated in 96-well culture plates (Corning, USA) at a density of 2×103 cells per well and cultured for 1, 3 and 7 days according to the different conditions of each group. HFF-1 cells and HaCaTs were cultured under the same conditions, but the initial number of cells was 1.5×103. Then, 100 µl of culture medium containing 10% CCK-8 was added to each well of the 96-well plate, and the plates were incubated for 2 h. The absorbance value of each sample was immediately measured at 450 nm by a microtiter plate reader (BioTek, Winooski, USA).
The migration of HUVECs, HFF-1 cells and HaCaTs was tested with a transwell assay (3422, Corning, USA). HUVECs, HFF-1 cells and HaCaTs at a density of 2×104 cells were inoculated in the upper chamber and cultured in 200 µl of serum-starved medium, whereas the lower chamber contained 500 µl of complete medium. The cells were fixed and stained for 10 min with 0.1% crystal violet after incubation for 24 h. The migrated cells were photographed by microscopy (Olympus IX 70, Tokyo, Japan) and counted by ImageJ.
Angiogenic characters
MatrigelTM (BD Bioscience) was thawed in advance in a 4°C refrigerator overnight. In precooled 24-well plates, 200 µl of Matrigel was added to each well, and the plates were then incubated at 37°C for 1 h. HUVECs that had been precultured for 48 h in medium under different conditions were digested with trypsin and counted. A total of 1x105 pretreated HUVECs were added to the 24-well plates containing Matrigel, and the samples continued to be cultured in the treated medium. The tube-forming ability of HUVECs was observed by microscopy (Olympus IX 70, Tokyo, Japan) after culture in a humidified 37°C/5% CO2 incubator for 8 h. Statistical analysis of the number of tubes in the microscope (Olympus IX 70, Tokyo, Japan) photos was carried out with Image J.
After culture in medium under the control condition or with 0.1 nM, 1 nM or 10 nM PTHrP-2 (n = 4) for 3 days, HUVECs and HFF-1 cells were fixed with 4% paraformaldehyde for 15 min and then washed with PBS three times. Next, the cells were permeabilized with 0.25% Triton X-100 for 15 min and then blocked with 3% bovine serum albumin (BSA) for 1 h. After washing with Phosphate Buffered Saline (PBS), we added anti-VEGF (1:200, ABclonal, China) and incubated with the cells in a 4°C refrigerator overnight; then, we added the secondary antibodies in darkness. One hour later, the cells were washed with PBS three times for 5 min each time. The cytoskeletons were then stained with 5 g/ml of phalloidin (1:200, Yeasen, China) at room temperature for 45 min. Then, the slides were washed with PBS three times for 5 min each time. After the final wash, the samples were stained by adding 4′,6-diamidino-2-phenylindole (DAPI, 1:200, Solarbio) in PBS for 10 min, followed by imaging. The cells were visualized using a confocal microscope (Leica, Solms, Germany).
The VEGF secretion from HUVECs was detected by an enzyme-linked immunosorbent assay (ELISA). A total of 1x105 HUVECs were seeded in medium under the control condition or with 0.1 nM, 1 nM or 10 nM PTHrP-2 (n=4) in 6-well plates. After the cells were cultured for 3 days, the supernatants of the samples were collected, and the contents of VEGF released from the samples were detected in strict accordance with the manufacturer's instructions using an ELISA kit (NeoBioscience, China).
Fibrogenic characters
Immunofluorescence (Anti-Collagen I, Abcam, UK) and enzyme-linked immunosorbent assay (ELISA) (Human Pro-Collagen I alpha 1 DuoSet ELISA, R&D systems, USA) were used to determine the type I collagen in HFF-1 cells.
Western blotting to evaluate angiogenic and fibrogenic characteristics
For Western blotting, exosomes or cells were lysed. The lysates were diluted with 5 × loading buffer at a ratio of 1:5 and heated at 95°C for 5 min. The protein extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Immobilon P, Millipore, Billerica, USA). The membranes were blocked with 5% nonfat milk or BSA for 1 h and then incubated with primary antibodies overnight at 4°C and with HRP-linked secondary antibodies for 1 h at room temperature. The protein bands were then visualized using an enhanced chemiluminescence (ECL) substrate kit (Merck Millipore, USA).
Exosome isolation
The HUVEC and HFF-1 exosomes were isolated by ultracentrifugation. In brief, when HUVEC and HFF-1 cultures reached 80% confluence, the culture medium was removed, and the cells were washed three times with PBS. Then, serum-free medium was used for culturing. PTHrP-2 was added to the medium of the PTHrP-2-treated group at this time. After 48 h of culture, conditioned media were collected and centrifuged at 300 × g for 10 min and 2000 × g for 15 min to remove dead cells and debris. The supernatants were then filtered via a 0.22-µm filter (Micropore) and centrifuged at 100000 × g for 1.5 h twice. Then, the pellets were resuspended in PBS.
Exosome characterization
The morphology of exosomes was identified by TEM (JEM-1400, JEOL, Japan). Western blotting analysis was used to verify the exosome markers Alix, TSG101 (Protein Tech, USA) and CD9 (Abcam, USA). DLS was applied to determine the exosome size distribution. Particle concentration, particle size and the video frame of exosomes were analyzed by a Flow NanoAnalyzer (FNA) (NanoFCM, China) and nanoparticle tracking analysis (NTA) (ZetaView PMX 110, Particle Metrix, Meerbusch, Germany). The protein concentration of exosomes was quantitatively detected by a BCA protein assay kit.
Exosome internalization
The purified exosomes were labeled with the red fluorescent dye PKH26 (Sigma-Aldrich, Germany) according to the manufacturer's protocol. Subsequently, PKH26-labeled exosomes were added to the medium and incubated with HUVECs, HFF-1 cells and HaCaTs for 24 h. Afterwards, the cells were fixed and washed with PBS 3 times and then blocked with QuickBlock™ Blocking Buffer for Immunol Staining (Beyotime, China). The cytoskeleton was then exposed to 5 g/ml of phalloidin (1:200, Yeasen) at room temperature for 45 min. Then, the slides were washed with PBS three times for 5 min each time. After the final wash, the samples were stained by adding DAPI (1:200, Solarbio) in PBS for 10 min and then imaged. The cells were visualized using a confocal microscope (Leica, Solms, Germany).
Exosome-mediated intercellular communication
Exosomes were extracted from HUVECs and HFF-1 cells in the PTHrP-2-treated and untreated groups using the method described above. Exosomes from the treated groups (PTHrP-2-HUVEC-Exos and PTHrP-2-HFF-1-Exos) and untreated groups (HUVEC-Exos and HFF-1-Exos) were cultured together with HUVECs and HFF-1 cells, and the proliferation, migration and tube formation experiments were performed as described above.
Effects of PTHrP-2-treated-exosomes on HaCaTs
The exosomes extracted above were co-cultured with HaCaTs to evaluate the proliferation and migration ability of HaCaTs according to the methods described above. According to the characterization results of HaCaTs, the mechanisms were explored by Western blot.
In vivo validation of PTHrP-2-HUVEC-Exos
The effect of PTHrP-2-HUVEC-Exos in vitro was verified by subcutaneous injection in diabetic rat wounds. Histopathological methods were used to analyze the rats 7 days after operation. HE staining, Masson staining, CD31 immunohistochemical staining and CD31/α-SMA dual immunofluorescence staining experiments were carried out according to the methods described above.
Statistical analysis
All experiments, both in vitro and in vivo, were repeated at least three times. Data were representative of these experiments and were shown as the mean ± standard deviation (SD). The means of multiple groups were compared with one-way analysis of variance (ANOVA). The independent sample test was used to compare means between two groups. Statistical analysis was conducted using GraphPad Prism software, and P < 0.05 was considered statistically significant.
{kind=link}