Cell culture and iPSC differentiation
Control (CS06iCTR), PSEN1 (CS40iFAD) and PSEN2 (CS08iFAD) iPSC lines used in this study were acquired from the Cedars-Sinai iPSC core (Los Angeles, CA). The PSEN1 iPSC line was isolated from a 56-year old Caucasian male diagnosed with memory impairment and harbors an Ala246Glu mutation. The PSEN2 iPSC line was isolated from an 81-year-old Caucasian female diagnosed with progressive dementia and harbors an Asn141Ile mutation.
Undifferentiated iPSCs were maintained in hESC-grade Matrigel® (Corning, Corning, NY) in presence of Essential 8 (E8) medium (Life Technologies, ThermoFisher, Waltham, MA) as previously described (24). iPSC differentiation into BMECs occurred following the differentiation protocol previously published by our lab (24). Briefly, cells were maintained in E8 for 5 days prior to differentiation, followed by 6 days in unconditioned medium [UM: Dulbecco’s modified Eagle’s medium/F12 with 15 mM HEPES (ThermoFisher), 20% knockout serum replacement (ThermoFisher), 1% non-essential amino acids (ThermoFisher), 0.5% Glutamax (ThermoFisher), and 0.1 mM b-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA)] and 2 days in EC+/+ [EC medium (ThermoFisher) supplemented with 1% platelet-poor derived serum (PDS, Alfa-Aesar, ThermoFisher, Haverhill, MA, USA), 20 ng/mL human recombinant basic fibroblast growth factor (Tocris, Abingdon, UK), and 10 µM retinoic acid (Sigma-Aldrich)]. At day 8 of differentiation, cells were enzymatically dissociated (Accutase®, Corning) and seeded on tissue culture plastic surfaces (TCPS) coated with collagen (isolated from human placenta, Sigma-Aldrich)/fibronectin (bovine plasma, Sigma-Aldrich) at concentrations of 80 µg/cm2 and 20 µg/cm2 respectively. At day 9 of differentiation, iPSC-derived BMECs were maintained in EC-/- medium [EC medium supplemented with 1% PDS] for 24 hours. Experiments were conducted at day 10 of differentiation.
Immunofluorescence
Cells were quickly washed with ice-cold PBS and fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA, USA) and blocked for 30 minutes at room temperature (RT) in presence of PBS supplemented with 10% goat serum (ThermoFisher) supplemented with 0.2% Triton-X100 (Sigma). Cells were incubated overnight at 4ºC in primary antibodies targeting BCRP (1:100, Millipore, RRID: AB_11213795), claudin-5 (1:100, Life Technologies, RRID: AB_2533200), GLUT1 (1:100, ThermoFisher, AB_10979643), GLUT3 (1:100, ThermoFisher, AB_2809974), GLUT4 (1:100, ThermoFisher, AB_11153908), MRP1 (1:100, Millipore, RRID: AB_2143819), occludin (1:100, Life Technologies, AB_2533101), P-gp (1:50, ThermoFisher, AB_1233253) and ZO1 (1:100, RRID:AB_2533938) diluted in 10% goat serum (PBSG). Primary antibodies detection occurred by incubation with goat-anti mouse Alexa Fluor® 555-conjugated secondary antibody (Life Technologies) for 1 hour at room temperature. Cells were observed at 200X magnification (20X long-distance dry objective) and acquired using a Leica DMi-8 inverted epifluorescence microscope (Leica Microsystems, Wetzlar, Germany). Background fluorescence was subtracted from unlabeled cells incubated with the secondary antibody only. Exposure time was set on the control iPSC-BMECs for each experiment and each protein of interest while maintained constant through the acquisition of other pictures in the PSEN1 and PSEN2 groups. Images were processed using ImageJ (Image J, NIH, Bethesda, MD). The average relative fluorescence was calculated by measuring the mean fluorescence from five microscopic fields (four cardinal direction and center of a well), and quantified using the built-in function in ImageJ. The average relative fluorescence was obtained for three independent biological replicates (three independent iPSC differentiation passages).
TEER and permeability experiments
Barrier tightness was measured by assessing both transcellular electrical resistance (TEER) and fluorescein permeability (paracellular tracer). TEER was measured using an EVOHM STX2 chopstick electrode (World Precision Instruments, Sarasota, FL, USA). For each experiment, three measurements were performed for each insert, and the average resistance obtained was used to determine barrier function. Fluorescein permeability was assessed by incubating 10 µM sodium fluorescein (Sigma-Aldrich) in the donor (apical) chamber, with sampling in the donor (basolateral) chamber every 15 minutes for up to 60 minutes. Fluorescein permeability (Pe) was calculated using the clearance slopes obtained by extrapolation using the following formula:

Pt and Pf indicative of the clearance slopes of samples and blank (empty coated) filters, and S indicative of the insert surface area (cm2).
Drug uptake assay
Cells were incubated in the presence of 10µM Rhodamine 123 (P-gp substrate, Sigma), FL-BOPIDY (BCRP substrate, Sigma) or CM-DCFDA (MRP substrate, Sigma) for 1 hour at 37ºC followed by cell lysis using RIPA buffer (ThermoFisher). For assessing the contribution of efflux pump in the drug uptake, cells were pre-incubated for 1 hour in presence of 5µM cyclosporine A (CsA, P-gp inhibitor, Sigma), 1µM Ko143 (BCRP inhibitor, Sigma) or 10µM MK571 (MRPs inhibitor, Sigma) and maintained during the incubation with drug efflux substrate. Following incubation, cells were briefly washed with ice-cold PBS and lysed with RIPA buffer. Fluorescence in cell lysates was assessed using a SynergyMX2 ELISA plate reader (Bio-Tek, Winooski, VT, USA). Relative fluorescence units (RFU) were normalized against the total protein content and the protein levels were determined by bicinchoninic acid assay (BCA, ThermoFisher). Fluorescence values (expressed as relative fluorescence unit or RFU) obtained from cell lysates in the absence of inhibitor (named as controls) were normalized to the protein content and expressed as RFU/µg protein.
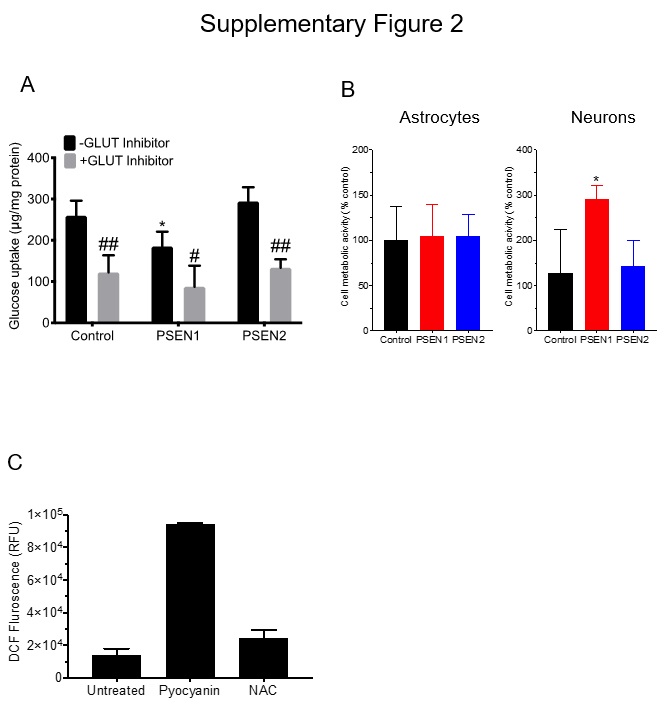
Glucose uptake assay
Cells were incubated in presence of [14C]-D-glucose (0.4µCi/mL) for 1 hour at 37ºC.
Following incubation, cells were briefly washed with ice-cold PBS and lysed with RIPA buffer. In experiments involving GLUT1 inhibition, cells were pre-incubated in presence of 10µM glucose transporter inhibitor II (Millipore-Sigma, Danvers, MA) for 1-hour prior incubation with glucose. Radioactivity in cell lysates was assessed using liquid scintillation cocktail (Scintisafe® 30%, ThermoFisher) and quantified with a Beckman-Coulter LS6500 (Beckman-Coulter, Brea, CA). Glucose uptake levels were normalized by the total amount of protein in samples.
Glycolytic flux and mitochondrial analysis
Glycolytic flux analysis was assessed using a Seahorse XFe-24 cell flux analyzer (Agilent Technologies, Santa Clara, CA). Cells were seeded at a density of 5 x 104 cells/well on custom-designed 24-well plates (Agilent Technologies) at day 8 of differentiation and allowed to grow for 48 hours. On the day of experiments, cell medium was replaced by glucose-free medium provided with the glycolytic stress test kit (Agilent) for 2 hours prior experiment. Cell medium was replaced once with glucose-free medium and initiated measurement of both the extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR). At 20 minutes of incubation, 10mM D-glucose was added in the incubation chamber, followed by the addition of 1µM of oligomycin at 40 minutes and finally addition of 100mM 2-deoxy-D-glucose (2-DG) at 60 minutes timepoint, with measurements occurring until the 90th minute timepoint. The cell energetic profile was determined at the 3rd timepoint by plotting the ECAR and OCR values for each well and delimited using the quadrants established by the manufacturer. The assessment of the glycolytic parameters was performed by the Seahorse Wave software data analyzer (Agilent), using the ECAR values reported during the experiments. Briefly, the glycolysis parameter was obtained by measuring changes in the ECAR following the addition of D-glucose, whereas the glycolytic reserve was determined by measuring the ECAR values following the oligomycin treatment. The glycolytic capacity was determined by subtraction of the glycolytic reserve minus the glycolysis. Finally, the non-glycolytic acidity was determined from the ECAR values following the 2-DG treatment.
In mitochondrial stress assays, changes in oxygen consumption rates were accomplished using the Mito Stress Test kit assay. Cells were maintained in assay medium (DMEM) for 1 hour at 37ºC (0% CO2) prior the performance of the assay as recommended per the manufacturer. The assay was initiated by measuring changes in the OCR values for 20 minutes to determine the basal respiration, followed by treatment with 1.5µM oligomycin (complex V ATP synthase inhibitor). This step allows us to the ATP-linked respiration (by measuring the difference in OCR before and after oligomycin treatment). Following treatment with oligomycin, cells are treated with 0.5µM FCCP (uncoupling agent) and allows the measurement of the spare capacity (OCR after FCCP treatment – basal respiration). Finally, 0.5µM rotenone/antimycin A (R/AA, complex I and III) allows the measurement of the maximal respiration (difference in OCR before and after R/AA treatment), calculation of the non-mitochondrial oxygen consumption (OCR measurement at the last timepoint) and proton leak (OCR following oligomycin treatment – OCR at last timepoint).
JC-1 flow cytometry and live imaging
At day 10 of differentiation, cells were enzymatically dissociated with Accutase®. Cells were resuspended and centrifuged and resuspended in medium containing 5µM JC-1 dye (ThermoFisher) for 30 minutes at 37ºC. In experiments involving FCCP treatment, cells were simultaneously treated with 50nM FCCP. Following incubation with JC-1 dye, cells were washed by centrifugation and resuspension in 200µL PBS for flow cytometry analysis. In experiments involving live imaging, cells were incubated in presence of 100µL of JC-1 reagent (Cayman Chemicals, Ann Arbor, MI) and incubated for 20 minutes at 37ºC/5% CO2.
Under physiological condition, a high mitochondrial membrane potential results in the formation of “J-aggregates”, which accumulates within the mitochondria, while emitting a fluorescence around 590nm (red). Under mitochondrial stress condition, JC-1 remains as monomers and emit a fluorescence around 530nm (green). Live cells were observed under the Leica DMi-8 microscope, with the use of the 485/535nm for impaired cells and 540/570nm for healthy cells to detect JC-1 aggregates, whereas cell events in the flow cytometry were counted at PMTs capturing fluorescence signals in the fluorescein isothiocyanate (FITC) and phycoerythrin (PE) emission ranges respectively. The fluorometric ratio of JC-1 was used as an indicator of mitochondrial dysfunction by assessing changes in mitochondrial membrane potential. In experiments involving FCCP treatment (an uncoupling agent disrupting the mitochondrial oxidative phosphorylation), cells were simultaneously treated with 50nM FCCP for 1-hour prior incubation with JC-1 dye.
Acridine orange (AO) flow cytometry
At day 10 of differentiation, cells were maintained for 24 hours in EC-/- medium or in serum-free EC medium to induce serum starvation. Following such treatment, cells were enzymatically dissociated with Accutase®. Cells were resuspended and centrifuged and resuspended in medium containing 1µg/mL acridine orange (AO, Sigma-Aldrich) dissolved in PBS and allowed to stain for 15 minutes, following the protocol of Thome and colleagues (25). Fluorescence detection in samples was performed using a FACSVerse® flow cytometer (BD Biosciences, San Jose, CA). Fluorescence PMTs were calibrated on unstained cells and set for the remaining of the experiments. Quadrants were set on control cells, with events expected to occur as FITChigh and PEhigh.
Under physiological condition, AO accumulates inside acidic vesicular organelles (AVOs, such as autophagic lysosomes) and emit a green fluorescence under such condition. However, in late-stage autophagy, AO can dimerize and shift its emission range into a red fluorescence. Therefore, the quantification of both AO monomer fluorescence in the green (FITC) and red (PE) channels are indicative of a functional basal autophagy and presence of AVOs. Induction of autophagy by serum starvation will result in an increase in such dimerization process and ultimately a shift in the emission pattern with a decrease in green fluorescence and an increase in red fluorescence.
Lysosensor(R) live imaging
Live cells were incubated in presence of 1µM Lysosensor-Green DND 189 for 5 minutes, followed by a brief wash with ice-cold PBS and fixation with 4% paraformaldehyde. Cells were counterstained with 300nM DAPI solution and immediately processed for imaging under the Leica DMi-8 inverted fluorescence microscope at 20X.
Radical oxygen species assays
At day 10 of differentiation, intracellular radical oxygen species (ROS) was assessed using both a qualitative and quantitative approach.
The qualitative approach in assessing ROS production was performed using CellROX(R) assay (ThermoFisher Scientific). CellROX(R) reagent was added to live cells at a final concentration of 5µM to the medium and incubated for 30 minutes at 37°C/5% CO2. The medium was removed, and cells were quickly washed with PBS. Cells were observed at 20X magnification, and images were acquired using a Leica DMi-8 inverted fluorescence microscope (Leica Microsystems). The quantitative approach in assessing ROS production was performed using a DCFDA-based cell kit (Cayman Chemicals), following the instruction provided by the vendor. DCFDA fluorescence was measured using SynergyMX2 ELISA plate reader (Bio-Tek). DCFDA fluorescence intensity in CTR90F BMECs was used as a baseline value for comparison to the PSEN BMECs.
Cell viability
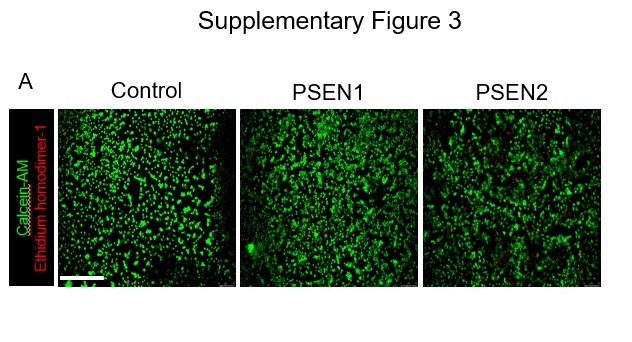
To study the effect of mutation on cell viability, a LIVE/DEAD™ Viability/Cytotoxicity Kit (Invitrogen) was used. A calcein-AM/ethidium homodimer-1 (EthD-1) assay was performed based on the manufacturer’s protocol. Briefly, cells were stained by a dye solution consisted of 0.5ul of Calcein AM and 0.5ul of ethidium homodimer-1 per ml PBS. Cells were observed after 20min of incubation at 20X magnification, and fluorescence was acquired using a Leica DMi-8 inverted fluorescence microscope (Leica Microsystems, Wetzlar, Germany). Ethidium homodimer-1 enters into cells with compromised cell membranes and subsequently intercalates with nucleic acids. Thus, dead cells have red fluorescence. Calcein-AM penetrates living cell membranes and produces green fluorescence by cleavage of cytoplasmic esterase.
Ab1-40 and Ab1-42 ELISA
Cell conditioned medium from BMECs monolayers grown at Day 10 were collected and immediately frozen at -80ºC. Ab1-40 and 1-42 levels in supernatants were measured using their respective ELISA Quantikine(R) kits (R&D Systems, Minneapolis, MN).
In experiments involving cell homogenates, cells were briefly washed with PBS followed by homogenization using 200µL RIPA buffer (ThermoFisher) supplemented with 1x protease inhibitors cocktail (ThermoFisher). Cells were maintained on ice for 10 minutes, followed by centrifugation at 14’000rpms for 10 minutes at 4ºC. The supernatant of such lysates was collected and immediately frozen at -80ºC pending analysis. Cell lysates protein concentrations were measured using BCA assay and normalized (using the diluent provided by the kit) to achieve a final protein concentration of 1µg total protein/µL if necessary. 200µL of such normalized samples were loaded onto the ELISA plate.
Statistics
Data are represented as mean ± S.D. from at least three independent experiments. Statistical analysis was performed using one-way or two-way analysis of the variance (ANOVA) using parametric tests. Group comparisons were performed against the control iPSC group, both in one-way and two-way ANOVA post-hoc analysis. Statistical analysis was performed using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA). A P-value lesser than 0.05 (P<0.05) was considered as indicative of a statistic difference between one or more groups.
{kind=link}
{kind=link}
{kind=link}