Reaction optimization. Initially, the alkyne-containing 1,3-dicarbonyl diazo compound 1a and styrene 2a were chosen as the model substrates to optimize the reaction conditions (Table 1). A variety of metal catalysts, such as RhI-, RhII-, CuII-, Pd0-, and AgI-complexes were examined in dichloroethane (DCE) at different temperatures, which all led to the tricyclic product 3’ in high to excellent yields rather than the desired tetracyclic product 3 (entries 1-5). Given the fact that the formation of 3’ with these catalysts should go through a carbene/alkyne metathesis process (CAM)52-61, the gold-complexes, which have shown unique ability to selectively activate alkyne species with the pendant diazo group serves as a latent functionality40-42,62, were then evaluated. Due to the competition between ligand and carbene for the contribution of electron density of gold center, the ligands of the gold catalysts have a significant influence on the bonding and reactivity of corresponding gold-complex intermediates29-34, and we have observed these dramatic influences in the outcome of the following optimization. The gold catalysts with trialkylphosphines as the ligands could produce mainly the polycarbocyclic product 3 in moderate conversions (entries 6-8, 29%-36% yields), whereas, the triarylphosphines showed relative lower reactivity (27%-39% conversions), preferring to form the intramolecular cyclization product 3’ (entries 9-11). Further investigation of phosphine ligands with structural and electronic diversity implied that ligands bearing electron-donating substituents (entries 12 vs 13) and with appropriate steric hindrance (entry 14 vs entries 12, 16, and 17) gave better results, affording 3 in 90% NMR yield when JohnPhos (L3) was used as the ligand (entry 14, 84% isolated yield). A comparably good result was obtained by switching the counter anion of gold catalyst from SbF6− to NTf2− (entry 15). Based on these results, we set out to explore a statistical regression approach to interpretation and prediction of ligand effects. The calculated Au-Cl bond distance, which has been disclosed by Fey and co-workers63, might provide such a platform to quantify the steric and electronic properties of these ligands64,65. Our optimization results have shown good correlation with the calculated parameters of the Au-Cl bond distance of gold-complexes with corresponding ligands (see Supplementary Fig. 1 for detail). Moreover, these results bring us to predict that electron-donating substituents with moderate steric hindrance on the phosphine ligand might further improve the yield. Thus, the ligand (Me2N)3P, which is similar to triisopropylphosphine (entry 8), but is much more flexible due to the additional freedom of nitrogen inversion and the three amino groups offer complementary donor functions66,67, was introduced. Gratifyingly, this ligand proved to the most effective one, delivering the desired product 3 in 89% isolated yield (entry 18).
Substrate scope. Under the optimized reaction conditions, the scope of this gold-catalyzed [4+2]-cycloaddition with respect to the 1,3-dicarbonyl diazo compound 1 in combination with styrene 2a was examined (Fig. 2). The substitutions on the aryl linkage (Ar1), including fluoro on the different positions (4-7), methyl (8), and methoxy (9) groups did not obviously affect the reactivity, and 82%-93% isolated yield was obtained in these cases. The diazo compound with naphthyl group as the linkage provided the pentacyclic product 10 in 53% yield. Then, the nature of the alkyne terminus was investigated (Ar2). The steric hindrance resulting from the ortho- and meta-methyl substituents on the phenyl ring did not impact the reactivity a lot, delivering corresponding products 11 and 12 in 75% and 95% yield, respectively. Other diazo derivatives, containing different substituents on the para-position of the aryl ring, performed well under these conditions (13-16), although low yields were obtained in the halogen-substituted cases. This may due to the lower nucleophilicity of these aromatic rings. Naphthyl- and thienyl-alkynes reacted effectively under gold-catalysis, offering the corresponding polycyclic products all in high yields (17-20). The installation of cyclohexenyl group proximal to the alkyne motif instead of aryl led to the product 21 as two isomers in 33% and 50% yields, respectively, and with 21b in 1.7:1 dr.
To further explore the substrate scope of this gold-catalyzed [4+2]-cycloaddition, we next examined a variety of olefins 2 (Fig. 3). The electronic effects and the position of the substituent groups on the phenyl ring of the styrene had little influence; substrates containing bromo, chloro, fluoro, trifluoromethyl, methyl, tert-butyl, and methoxy groups effectively reacted with 1l to form the tetracyclic products 22-30 in synthetically useful to high yields. Relatively high yields were obtained when an electron-withdrawing substituent was incorporated in the arene. Olefins with bulky ortho-substituent also tolerated in this reaction, selectively affording the corresponding products 31-35 as signal diastereomer in 49%-94% yields. Even 1,2-divinylbenzene could be used, interestingly only the mono-cycloaddition product 36 was generated in 81% yield as a mixture of two diastereomers. The diastereomers resulted from the additionally formed axial chirality due to the hindered rotation of the ortho-substituted aryl group in these products. Despite the inefficiency of the reaction with internal and alkyl alkenes, which only generated the intramolecular cyclization byproducts from 1l, the disubstituted terminal alkene, 1-methyl-1-phenylethene, worked well, leading to the cycloadduct 35 in 76% yield. The structures of 23 and 34 were confirmed by single-crystal X-ray diffraction analysis.
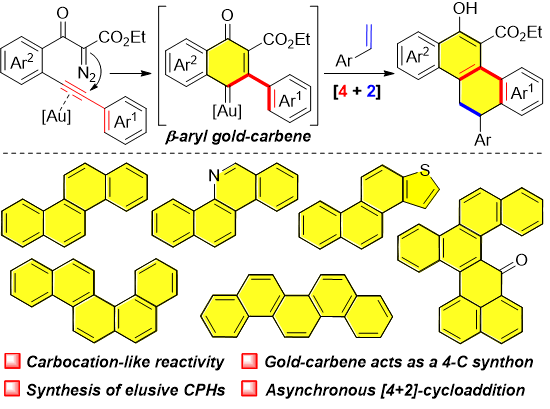
Development of the oxidation procedure for polycyclic aromatic hydrocarbons (PAHs) With these polycarbocyclic products in hand, we then achieved their transformation to the corresponding CPHs. The aromatization occurred smoothly in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) under mild and neutral conditions (Fig. 4). Various functional groups, such as phenolic hydroxyl, ester, methoxy, halogen, alkyl, and aryl were well tolerated, assembling multiple substituted CPHs 38-53 in high to excellent yields. In addition to the chrysene derivatives, a few of elusive CPHs, including picene (46, 86%), benzo[c]chrysene (47, 98%), and phenanthro[2,1-b]thiophene (48, 84%) were also readily prepared in high yields. It could be envisioned that additional different types of polyaromatic hydrocarbons with structural diversity would become accessible with this method by the manipulation of the structure of the substrates or via further synthetic transformations.
Optical properties. After the construction of these elusive p-conjugated polycyclic hydrocarbons (CPHs), we then investigated the optical properties of representative analogous in DMSO (Fig. 5). The absorption spectra of tested PAHs displayed the λmax in the range of 402-421 nm. For the fluorescence spectra, compounds 43, 45, 50, and 51 exhibited sky-blue lights with similar peaks at around 490 nm; whereas, the five-fused aromatic product 46 showed green light with a maximum emission of 550 nm due to the extension of the p-conjugated system.
Mechanistic studies. Mechanistic experiments were performed to gain insights into the reaction pathway of this transformation (Fig. 6). To verify the existence of the on-ring β-aryl gold-carbene intermediate, the interception reaction with 1l in the presence of diphenyl sulfoxide (1.5 equiv), instead of styrene, was carried out under standard conditions, and the corresponding cyclic ketone product 54 was isolated in 84% yield (Fig. 6a). These results are also consistent with a direct 6-endo-dig diazo-yne carbocyclization process for the generation of this on-ring carbene intermediate, otherwise, the linear ketone product that furnished via direct carbonylation of the diazo group might be observed43. Evidence for the carbocation-like reactivity of this generated gold-complex was verified by the reactions of 1l with two disubstituted terminal alkenes, 1,1-diphenylethylene and 1-phenyl-1-trimethylsiloxyethylene, delivering the coupling-type and addition-type products 55 and 56 in 71% and 80% yields, respectively (Fig. 6b and 6c). Moreover, a non-concerted, step-wise mechanism of the cyclization process was well supported by the interception reaction with external alcohol. The identifiable three-component product 57 was isolated in 79% yield when the reaction was carried out in the presence of o-bromobenzyl alcohol (Fig. 6d). The comparison reaction with 1,3-dicarbonyl diazo compound 1aa without the alkyne species turned out that only very slowly decomposition of the diazo compound was observed under the current conditions (Fig. 6e). The 31P NMR analysis results by mixing the gold catalyst (5.0 mol%) with 1l (0.02 mmol) in CDCl3 at 20 °C suggested that, rather than direct decomposition of the diazo species59-61, the formation of a relative stable Au-alkyne complex is favorable under these conditions (Fig. 6f, and see Supplementary Fig. 2 for detail). In addition, this protocol could also be applied for the preparation of benzo[c]phenanthridine 58 and 6H-dibenzo[c,h]chromene 59 from corresponding materials in 91% and 94% yields, respectively (Fig. 7a and 7b). All these results well rationalized the reaction mechanism, and also underlined the synthetic potential of this method in diversity-oriented synthesis.
Based on the above studies and the reported literature40-47, a possible reaction mechanism is depicted in Fig. 8. Initially, the gold-promoted 6-endo-dig carbocyclization of 1 formed intermediate B via A, which further led to the key intermediate vinyl gold carbene C after the extrusion of N2. The resonance phenomenon of this gold-complex between carbene (C) and carbocation (C’) forms, which mainly depends on the ligands of the gold catalyst, might be existing. In this case, carbocation-like intermediate C’ has been suggested based on the observations of the control experiments (Fig. 6). Subsequently, this gold intermediate reacted with an external alkene to form the carbocation intermediate D, followed by a Friedel-Crafts-type cyclization, furnishing the corresponding polycyclic products, and regenerating the gold catalyst. It is worth noting that this is the first example of an in situ-generated β-aryl gold carbene serving as a 4-C synthon in a cycloaddition reaction.
Applications. To demonstrate the utility of the current method, we performed the reaction on a gram scale (Fig. 9, 4.0 mmol), providing 1.48 g of 14 in 87% yield. Then, the cycloadduct 14 was subjected to further transformations. Sulfonylation of the phenolic hydroxyl group with trifluoromethanesulfonic anhydride (Tf2O) led to the coupling precursor 60 in quantitative yield. The following Sonogashira and Suzuki coupling reactions with terminal alkyne and naphthylboronic acid gave 61 and 62 in 93% and 98% yields, respectively. Oxidation of 62 with DDQ followed by an acid-promoted Friedel-Crafts-type intramolecular cyclization delivered the polycyclic hydrocarbon 63 in a total 82% yield for the two steps.
{kind=link}